Generalidad
La enfermedad de Wilson, también llamada degeneración hepatolenticular, es un trastorno genético poco común caracterizado por una acumulación de cobre en los tejidos y órganos del cuerpo.

Es una enfermedad mortal, por lo que se necesita un tratamiento terapéutico que elimine el cobre de los tejidos y evite su acumulación.
También lo es la enfermedad de Wilson
La enfermedad de Wilson, también conocida como degeneración hepatolenticular, es una enfermedad genética hereditaria que resulta en una acumulación excesiva de cobre en ciertos órganos y tejidos.
Es una enfermedad rara que afecta a 1 de cada 30.000 personas.
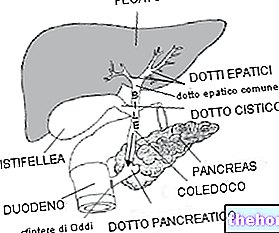
La acumulación de cobre se debe a un defecto en su metabolismo, de hecho, el cobre absorbido con la dieta no se excreta adecuadamente, por lo que permanece en el organismo y se deposita principalmente en:
- hígado;
- cerebro.
Y en menor medida, también en:
- córnea;
- riñones
- otras telas.
Cantidades excesivas de cobre en estas áreas causan daño a las células. Los efectos más graves se encuentran en el hígado y el cerebro. En el cerebro, es el núcleo lenticular el que sufre las mayores consecuencias: de ahí el nombre alternativo de degeneración hepatolenticular.
Causas
La causa de la enfermedad de Wilson es una "alteración del gen ATP7B, ubicado en el cromosoma 13, que ya no realiza su función normal".
La función del gen ATP7B es promover la excreción, a través de la bilis, del exceso de cobre contenido en las células. Cuando el ATP7B falla, el cobre se acumula en dosis tan masivas que escapa de las células y fluye hacia la sangre. el cobre llega a los distintos tejidos del cuerpo.
Patogénesis
El cobre se consume en la dieta. Su absorción ocurre en el intestino: aquí se une a la albúmina (una proteína plasmática) y llega al hígado. En este punto:
en un individuo sano:
- ATP7B promueve la unión entre el cobre y la ceruloplasmina. La ceruloplasmina es una proteína plasmática utilizada para el transporte y excreción de cobre.
Sin embargo, en un individuo con enfermedad de Wilson:
- ATP7B no funciona. Por tanto, no favorece la unión entre el cobre y la ceruloplasmina.
- El cobre permanece unido a la albúmina, no se excreta y se acumula en las células del hígado.
- Las células del hígado saturan cualquier capacidad de almacenamiento de cobre dentro de ellas.
- Por tanto, el complejo cobre-albúmina está en exceso. Por tanto, se escapa de los hepatocitos y entra en la sangre.
- A través de la sangre, el cobre llega a otros tejidos del cuerpo.
El primer órgano que paga las consecuencias es, por tanto, el hígado; siguen el cerebro, los riñones y la córnea.

¿Por qué el cobre se extiende a los textiles?
En las personas con enfermedad de Wilson, el cobre circula en la sangre unido a la albúmina. El enlace cobre-albúmina es mucho más lábil que el que existe entre el cobre y la ceruloplasmina. De hecho, hay poca afinidad entre las dos primeras c ". Cuando el cobre complejado con la albúmina llega a los tejidos y los distintos órganos, encuentra sustancias por las que tiene mayor afinidad y se une a ellas. Las consecuencias son dos:
- Los tejidos y órganos están enriquecidos con cobre.
- La concentración de cobre (cupremia) en la sangre disminuye.

De Wikipedia: la madre y el padre tienen un alelo mutado. Debido a la naturaleza recesiva de este alelo, no manifiestan ninguna enfermedad, pero son portadores sanos. Ambos padres pueden transmitir un alelo mutado a un niño cada uno. En este caso, el niño será homocigoto para el alelo dado y manifestará la enfermedad. En todos los demás casos, la presencia de uno o ambos alelos sanos no causa ninguna alteración.
Herencia
La enfermedad de Wilson es una enfermedad hereditaria autosómica recesiva.
- Autosómico, porque el gen ATP7B se encuentra en el cromosoma 13, un cromosoma no sexual.
- Recesivo, porque el alelo mutado, que determina la enfermedad, es recesivo en comparación con el sano. Para enfermarse, un individuo debe tener ambos alelos mutados. De hecho, un solo alelo mutado no es suficiente para causar la enfermedad. Uno de cada 100 people circa lleva un alelo ATP7B alterado. La figura explica claramente este concepto.
Síntomas
Para más información: Síntomas de la enfermedad de Wilson
Aunque se trata de una enfermedad genética hereditaria, en los primeros años de edad no hay alteraciones. Los primeros síntomas, basados en el hígado, aparecen alrededor de los 6 años. Este suele ser el tiempo mínimo que tarda el cobre en acumularse en cantidades dañinas. En algunos casos, el inicio puede ocurrir al final de la adolescencia o incluso alrededor de los 30 a 40. Con el tiempo, los trastornos también aparecen en otros tejidos.
Síntomas del hígado
El hígado es el primer órgano afectado, porque es el primer distrito al que llega el cobre absorbido de la dieta. La salud del hígado se deteriora progresivamente. La evolución generalmente comienza en la adolescencia y sigue el siguiente curso:
- Hepatitis.
- Cirrosis no grave.
- Cirrosis severa.
Se crea una condición definida por el médico con el término insuficiencia hepática: el hígado ya no puede realizar sus funciones.
Los signos típicos de insuficiencia hepática son:
- Ictericia.
- Dolor abdominal.
- Él vomitó.
- Agrandamiento del hígado (hepatomegalia)
- Agrandamiento del bazo (esplenomegalia)
Sintomatología cerebral
El cobre solo llega al cerebro cuando el hígado ya no puede mantenerlo confinado a sus propias células.
Los depósitos en el cerebro causan daños neurológicos de diferente naturaleza:
- Dolencias fisicas.
- Temblores de las extremidades.
- Lentitud de movimiento.
- Dificultad para hablar (disartria).
- Dificultad para escribir.
- Dificultad para tragar (disfagia).
- Inestabilidad al caminar.
- Migraña.
- Epilepsia.
- Debilidad y rigidez muscular.
- Trastornos del comportamiento.
- Cambios de humor.
- Depresión.
- Incapacidad para concentrarse.
- Cambios de personalidad.
- Demencia.
Si el paciente no recibe tratamiento, el daño neurológico empeora cada vez más: el individuo se vuelve completamente dependiente de los demás, para alimentarse y moverse.
Otros tejidos

Además, el cobre también se puede depositar en los riñones. Se produce daño renal, lo que resulta en:
- Aminoaciduria. Presencia de aminoácidos en la orina.
- Glucosuria. Presencia de glucosa en la orina.
- Fosfaturia. Presencia de fósforo en la orina.
- Uricosuria. Presencia de ácido úrico en la orina.
- Calciuria. Presencia de calcio en la orina.
En condiciones normales, todas estas sustancias perdidas se reabsorberían. Por tanto, la acumulación renal de cobre altera la estructura y la reabsorción de sustancias aún útiles para el organismo.
Otros posibles síntomas de la enfermedad de Wilson son:
- Anemia.
- Pancreatitis
- Problemas menstruales.
- Aborto espontáneo.
- Osteoporosis prematura.
Diagnóstico
Si se sospecha la enfermedad de Wilson, las pruebas de diagnóstico útiles son:
- Análisis de sangre, para probar:
- Concentraciones de ceruloplasmina. Un nivel bajo, por debajo de 20 mg / 100 ml, es indicativo de la enfermedad. El valor normal es de 30 mg / 100 ml.
- La concentración de cobre (cupremia). Si es menos de lo normal, es indicativo de la enfermedad.
- Posible anemia hemolítica.
- Funciones hepáticas y renales, a través de sus respectivos marcadores (transaminasas, azotemia, etc.)
- Análisis de orina, para evaluar la cantidad de cobre presente (cupruria). Los niveles por encima de lo normal son indicativos de la enfermedad. Por lo general, las personas con la afección excretan alrededor de 100 μg de cobre en la orina cada 24 horas.
- Examen optométrico, para detectar la presencia del anillo Kaiser-Fleischer.
- Biopsia de hígado, para medir el contenido de cobre en las células del hígado. El nivel patológico de cobre es superior a 100 μg por gramo de hígado. También es útil para evaluar el estado de la cirrosis.
- Una resonancia magnética del cerebro, para evaluar la salud del núcleo lenticular, que recordamos es el área del cerebro afectada por la acumulación de cobre.
- Una prueba de ADN genético.
La presencia simultánea de:
- Anillo Kaiser-Fleischer.
- Signos de cirrosis hepática.
- Lesión del núcleo lenticular.
no dejes ninguna duda sobre el diagnóstico.
Parámetro medido
Cupremia
110 μg / ml
<100 μg / ml
Cupruria
100 μg / 24 horas
>> 100 μg / 24 h
Ceruloplasmina
30 mg / ml
<20 mg / ml
Terapia
Ver también: Medicamentos para la insuficiencia suprarrenal
Si no se trata, la enfermedad de Wilson es fatal. La muerte puede ocurrir incluso un par de años después de que aparezcan los primeros síntomas. El paciente está sujeto a un empeoramiento progresivo de su condición, se vuelve cada vez más dependiente de los demás y, en ausencia de un tratamiento específico, el daño hepático y cerebral puede ser irreversible.
La terapia consiste en:
- Reducir los depósitos de cobre en el hígado.
- Verifique la absorción de cobre en el intestino.
- Reducir la introducción de cobre ingerido con la dieta.
- Trasplante de hígado.
Reducir los depósitos de cobre
Es el paso más importante para salvar la vida del paciente. Se basa en la administración de:
- Penicilamina.
- Trientina.
La penicilamina es el fármaco de elección, su administración se realiza por vía oral y debe tomarse de por vida, representa un agente quelante capaz de secuestrar el exceso de cobre y conducirlo a los riñones para su excreción. Sin embargo, puede provocar efectos indeseables en el propio riñón. En estos casos, es recomendable interrumpir el tratamiento para resolver los problemas que han surgido y adoptar una alternativa basada en la trientina.
La trientina también es un agente quelante, se administra por vía oral y actúa como la penicilamina, no es tan eficaz, pero los efectos secundarios también son menores.
Compruebe la absorción intestinal de cobre.
Es posible reducir la absorción de cobre tomando zinc, lo que evita la acumulación de cobre en el hígado. Se recomienda la administración de zinc cuando la enfermedad de Wilson se encuentra en sus primeras etapas. Es decir, cuando el cobre aún no ha invadido los demás tejidos. La terapia es eficaz cuando se combina con el tratamiento con penicilamina.
Reducir la introducción de cobre
Se debe limitar el consumo de ciertos alimentos ricos en cobre, como:
- Nueces
- Hígado.
- Champiñones.
- Chocolate.
- Mariscos.
En general, la ingesta diaria de cobre en la dieta no debe exceder los 2 mg.
Trasplante de hígado
Esta es la terapia necesaria si:
- El daño hepático es irreversible. En este caso, hablamos de cirrosis severa.
- Los tratamientos anteriores han sido ineficaces.
Pronóstico
Cuanto antes se inicie la terapia, mejor será el pronóstico y la calidad de vida.
Intervenir tarde significa limitar y mejorar sólo parcialmente el daño hepático y cerebral debido al exceso de cobre, algunas funciones, de hecho, quedan irremediablemente comprometidas.
En casos graves, la única solución para un mejor pronóstico es un trasplante de hígado.
















-cos-cause-e-terapia.jpg)