Ingredientes activos: Bortezomib
VELCADE 3,5 mg polvo para solución inyectable.
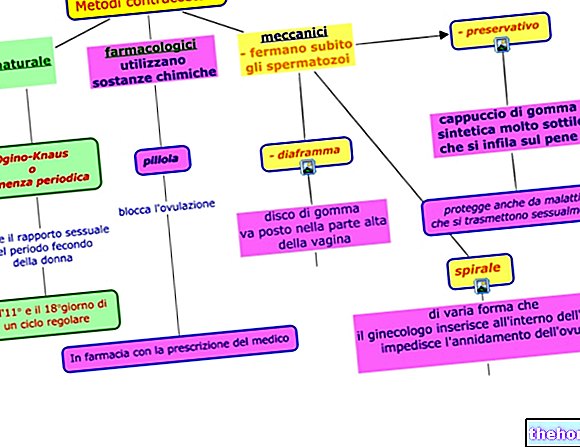
Indicaciones ¿Por qué se usa Velcade? ¿Para qué sirve?
VELCADE contiene el principio activo bortezomib, un denominado "inhibidor del proteasoma". Los proteosomas juegan un papel importante en el control de la función y el crecimiento celular. Al interferir con su función, bortezomib puede destruir las células cancerosas.
VELCADE se utiliza para tratar el mieloma múltiple (un tipo de neoplasia maligna de la médula ósea) en pacientes mayores de 18 años:
- solo o junto con los medicamentos doxorrubicina liposomal pegilada o dexametasona, para pacientes con empeoramiento (progresivo) de la enfermedad después de recibir al menos un tratamiento previo o en los que el trasplante de células madre sanguíneas ha fallado o no es factible
- en combinación con los medicamentos melfalán y prednisona para pacientes con enfermedades no tratadas previamente que no pueden recibir quimioterapia en dosis altas con trasplante de células madre sanguíneas.
- en combinación con dexametasona o dexametasona junto con talidomida, para pacientes con enfermedad no tratada previamente y antes de recibir quimioterapia de dosis alta con trasplante de células madre sanguíneas (tratamiento de inducción)
VELCADE se usa para tratar el linfoma de células del manto (un tipo de malignidad que afecta los ganglios linfáticos) en pacientes de 18 años de edad o mayores. En este caso, VELCADE se usa en combinación con los medicamentos rituximab, ciclofosfamida, doxorrubicina y prednisona, para pacientes con enfermedad no tratada previamente y para quienes el trasplante de células madre sanguíneas no es factible.
Contraindicaciones Cuándo no se debe usar Velcade
No use VELCADE
- si es alérgico al bortezomib, al boro oa cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si tiene problemas graves de pulmón o corazón.
Precauciones de uso Lo que necesita saber antes de tomar Velcade
Informe a su médico si tiene:
- bajo número de glóbulos rojos o glóbulos blancos
- problemas de sangrado y / o recuentos bajos de plaquetas en sangre
- diarrea, estreñimiento, náuseas o vómitos
- experiencias previas de desmayos, mareos o aturdimiento
- problemas de riñon
- problemas hepáticos de moderados a graves
- quejas previas como entumecimiento, hormigueo o dolor en las manos o los pies (neuropatía)
- trastorno del corazón o de la presión arterial
- dificultad para respirar o tos
- convulsiones
- herpes zóster (también ubicado alrededor de los ojos o que se extiende al resto del cuerpo)
- síntomas del síndrome de lisis tumoral como, por ejemplo, calambres musculares, debilidad muscular, confusión, alteración de la visión o pérdida de la visión y dificultad para respirar
- pérdida de memoria, dificultad para pensar, dificultad para caminar o pérdida de la visión. Estos pueden ser signos de una infección cerebral grave, y su médico puede indicarle más pruebas y controles.
Deberá realizarse análisis de sangre con regularidad antes y durante el tratamiento con VELCADE para comprobar constantemente los valores de sus células sanguíneas.
Si tiene linfoma de células del manto y le administran rituximab junto con VELCADE, debe informar a su médico:
- si cree que tiene hepatitis o la ha tenido en el pasado. En algunos casos, los pacientes que han tenido hepatitis B pueden tener un nuevo ataque de hepatitis, que puede ser mortal. Si ha tenido una infección por hepatitis B en el pasado, su médico deberá vigilarle de cerca para detectar signos y síntomas de hepatitis activa. B.
Lea los prospectos de todos los medicamentos que toma en combinación con VELCADE para obtener información sobre estos medicamentos antes de iniciar el tratamiento con VELCADE.
Cuando VELCADE se administre junto con el medicamento talidomida, preste especial atención a los consejos sobre las pruebas de embarazo y el programa de prevención del embarazo (ver "Embarazo y lactancia" en esta sección).
Niños y adolescentes
VELCADE no debe usarse en niños y adolescentes porque se desconoce cómo actúa el medicamento en estas personas.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Velcade?
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
En particular, informe a su médico si está utilizando medicamentos que contengan alguno de los siguientes principios activos:
- ketoconazol, utilizado para tratar infecciones por hongos
- ritonavir, utilizado para tratar la infección por VIH - rifampicina, un antibiótico utilizado para tratar infecciones bacterianas
- carbamazepina, fenitoína o fenobarbital, utilizados para tratar la epilepsia
- Hierba de San Juan (Hypericum perforatum), utilizada para tratar la depresión u otras afecciones.
- agentes antidiabéticos orales.
Advertencias Es importante saber que:
Embarazo y lactancia
No debe usar VELCADE si está embarazada a menos que sea claramente necesario.
Los hombres y mujeres en tratamiento con VELCADE deben utilizar métodos anticonceptivos eficaces durante y hasta 3 meses después del tratamiento. Si, a pesar de estas precauciones, queda embarazada, informe a su médico de inmediato.
No debe amamantar mientras toma VELCADE. Hable con su médico sobre el mejor momento para comenzar a amamantar nuevamente después de finalizar la terapia.
El medicamento talidomida causa defectos de nacimiento y muerte fetal. Cuando VELCADE se coadministra con talidomida, debe seguir el programa de prevención del embarazo con talidomida (ver el prospecto de talidomida).
Conducción y uso de máquinas
VELCADE puede provocar fatiga, mareos, desmayos o visión borrosa. No conduzca automóviles ni utilice máquinas si experimenta alguno de estos síntomas. También preste especial atención si estos efectos no ocurren.
Dosis, método y momento de administración Cómo utilizar Velcade: Posología
Su médico calculará la dosis de VELCADE en proporción a su altura y peso. La dosis inicial estándar de VELCADE es de 1,3 mg / m2 de superficie corporal dos veces por semana. Su médico puede cambiar la dosis y el número total de ciclos de tratamiento dependiendo de su respuesta al tratamiento, la aparición de ciertos efectos secundarios y sus condiciones generales de salud (por ejemplo, problemas hepáticos).
Mieloma múltiple en progresión
Cuando VELCADE se administra solo, recibirá 4 dosis de VELCADE por vía intravenosa o subcutánea los días 1, 4, 8 y 11. A esto le sigue un período de "descanso" de 10 días sin tratamiento.
Este período de 21 días (3 semanas) corresponde a un curso de tratamiento.
Puede recibir hasta 8 ciclos (24 semanas). También puede recibir VELCADE junto con los medicamentos dexametasona o doxorrubicina liposomal pegilada.
Cuando VELCADE se administra junto con doxorrubicina liposomal pegilada, recibirá un ciclo de 21 días de VELCADE intravenoso o subcutáneo y se administrarán 30 mg / m2 de doxorrubicina liposomal pegilada el día 4 del ciclo de tratamiento de 21 días de VELCADE en forma de perfusión. por vía intravenosa después de la inyección de VELCADE.
Puede recibir hasta 8 ciclos (24 semanas de tratamiento).
Cuando VELCADE se coadministra con dexametasona, recibirá un ciclo de tratamiento de 21 días con VELCADE intravenoso o subcutáneo y dexametasona oral a una dosis de 20 mg los días 1, 2, 4, 5, 8, 9, 11 y 12. del curso de 21 días de tratamiento con VELCADE.
Puede recibir hasta 8 ciclos (24 semanas de tratamiento).
Mieloma múltiple no tratado previamente
Si nunca antes ha recibido tratamiento para el mieloma múltiple y no es candidato para un trasplante de células madre sanguíneas, recibirá VELCADE junto con otros dos medicamentos: melfalán y prednisona.
En este caso, la duración de un curso de tratamiento es de 42 días (6 semanas). Recibirá 9 ciclos (54 semanas).
- En los ciclos 1-4, VELCADE se administra dos veces por semana los días 1, 4, 8, 11, 22, 25, 29 y 32.
- En los ciclos 5-9, VELCADE se administra una vez a la semana los días 1, 8, 22 y 29.
Melfalán (9 mg / m2) y prednisona (60 mg / m2) se administran por vía oral los días 1, 2, 3 y 4 de la primera semana de cada ciclo.
Si nunca antes ha recibido tratamiento por mieloma múltiple y es candidato para un trasplante de células madre sanguíneas, recibirá VELCADE por vía intravenosa o subcutánea junto con los medicamentos: dexametasona o dexametasona y talidomida, como tratamiento de inducción.
Cuando VELCADE se coadministra con dexametasona, recibirá un ciclo de tratamiento de 21 días con VELCADE intravenoso o subcutáneo y dexametasona 40 mg por vía oral los días 1, 2, 3, 4, 8, 9, 10 y 11 del ciclo. -Día de tratamiento con VELCADE.
Recibirá 4 ciclos (12 semanas de tratamiento).
Cuando VELCADE se administra junto con talidomida y dexametasona, la duración del ciclo de tratamiento es de 28 días (4 semanas).
La dexametasona 40 mg se administra por vía oral los días 1, 2, 3, 4, 8, 9, 10 y 11 del ciclo de tratamiento de 28 días con VELCADE y la talidomida se administra por vía oral a una dosis de 50 mg al día hasta el día 14 del primer día. ciclo y, si se tolera, la dosis de talidomida se aumenta a 100 mg los días 15-28 y posteriormente se puede aumentar hasta 200 mg por día a partir del segundo ciclo en adelante. Puede recibir hasta 6 ciclos (24 semanas de tratamiento).
Linfoma de células del manto no tratado previamente
Si nunca ha recibido un tratamiento específico para el linfoma de células del manto en el pasado, recibirá VELCADE por vía intravenosa o subcutánea junto con los medicamentos rituximab, ciclofosfamida, doxorrubicina y prednisona.
VELCADE se administra por vía intravenosa o subcutánea los días 1, 4, 8 y 11, seguido de un período de "descanso" sin tratamiento. La duración del curso de tratamiento es de 21 días (3 semanas).
Puede recibir hasta 8 ciclos de tratamiento (24 semanas).
Los siguientes medicamentos se administran el día 1 de cada ciclo de tratamiento de 21 días de VELCADE como perfusión intravenosa: rituximab a 375 mg / m2, ciclofosfamida a 750 mg / m2 y doxorrubicina a 50 mg / m2.
La prednisona se administra por vía oral a una dosis de 100 mg / m2 los días 1, 2, 3, 4 y 5 del ciclo de tratamiento con VELCADE.
Cómo se administra VELCADE
Este medicamento es para uso intravenoso o subcutáneo. VELCADE será administrado por un profesional sanitario con experiencia en el uso de medicamentos citotóxicos. El polvo de VELCADE debe disolverse antes de la administración. Esto lo hará un profesional sanitario. La solución resultante se inyectará rápidamente en una vena o por vía subcutánea. La inyección de L "en una vena es rápida, durante un período de 3 a 5 segundos. La inyección subcutánea se puede realizar en el muslo o en el abdomen.
Sobredosis Qué hacer si ha tomado demasiado Velcade
Como este medicamento lo administra su médico o enfermera, es poco probable que tome más de lo que debiera. En el caso poco probable de una sobredosis, su médico controlará los efectos secundarios.
Efectos secundarios ¿Cuáles son los efectos secundarios de Velcade?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Algunos de estos efectos pueden ser graves.
Si le administran VELCADE para el mieloma múltiple o el linfoma de células del manto, informe a su médico de inmediato si nota alguno de los siguientes síntomas:
- calambres musculares, debilidad muscular
- confusión, alteración o pérdida de la visión, ceguera, convulsiones, dolor de cabeza
- dificultad para respirar, hinchazón de los pies o cambios en la frecuencia cardíaca, presión arterial alta, cansancio, desmayos
- tos y dificultad para respirar u opresión en el pecho.
El tratamiento con VELCADE puede causar muy comúnmente una disminución en el número de glóbulos rojos y blancos y plaquetas de la sangre. Por lo tanto, deberá realizarse análisis de sangre con regularidad antes y durante el tratamiento con VELCADE, para controlar su recuento de células sanguíneas con regularidad. Puede experimentar una reducción en la cantidad de:
- plaquetas, que pueden hacerlo más propenso a sufrir hematomas o hemorragias sin una lesión perceptible (por ejemplo, hemorragia en los intestinos, estómago, boca y encías o hemorragia cerebral o hepática)
- glóbulos rojos, que pueden causar anemia, con síntomas como fatiga y palidez
- glóbulos blancos, que pueden hacerlo más susceptible a infecciones o síntomas similares a los de la gripe.
Si se le administra VELCADE para el tratamiento del mieloma múltiple, a continuación se enumeran los efectos secundarios que pueden ocurrir.
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
- Sensibilización, entumecimiento, hormigueo o sensación de ardor en la piel, o dolor en manos o pies, debido a daño en los nervios.
- Reducción del número de glóbulos rojos y / o blancos (ver arriba).
- Fiebre.
- Sensación de náuseas o vómitos, pérdida de apetito.
- Estreñimiento con o sin exceso de gases (puede ser severo).
- Diarrea: si esto ocurre es importante que beba mucha más agua de lo habitual. Su médico puede recetarle medicamentos para controlar la diarrea.
- Cansancio (fatiga), sensación de debilidad.
- Dolor muscular, dolor de huesos.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas)
- Presión arterial baja, caída repentina de la presión arterial al estar de pie que puede provocar desmayos.
- Aumento de la presión arterial.
- Función reducida de los riñones.
- Dolor de cabeza.
- Sensación de malestar general, dolor, mareos, aturdimiento, sensación de debilidad o pérdida del conocimiento.
- Escalofríos.
- Infecciones, que incluyen neumonía, infecciones respiratorias, bronquitis, infección por hongos, tos con flema, enfermedad similar a la gripe.
- Infección por herpes zóster (localizada, incluso alrededor de los ojos, o diseminada por todo el cuerpo).
- Dolor de pecho o dificultad para respirar durante la actividad física.
- Diferentes tipos de erupción cutánea (erupción).
- Picazón en la piel, bultos en la piel o piel seca.
- Enrojecimiento de la cara o pequeñas roturas en los capilares.
- Enrojecimiento de la piel.
- Deshidración.
- Acidez, hinchazón, eructos, gases, dolor de estómago, hemorragia intestinal o estomacal.
- Alteración de la función hepática.
- Irritación de la boca o labios, boca seca, úlceras bucales o dolor de garganta.
- Pérdida de peso, pérdida del gusto.
- Calambres musculares, espasmos musculares, debilidad muscular, dolor en brazos y piernas.
- Visión borrosa.
- Infección de la capa más externa de los ojos y la superficie interna de los párpados (conjuntivitis).
- Hemorragia nasal (sangrado).
- Alteraciones o problemas del sueño, sudoración, ansiedad, cambios de humor, estado de ánimo deprimido, inquietud o agitación, cambios en el estado mental, desorientación.
- Hinchazón del cuerpo, incluida la hinchazón alrededor de los ojos y otras partes del cuerpo.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
- Insuficiencia cardíaca, ataque cardíaco, dolor de pecho, malestar en el pecho, aumento o disminución de la frecuencia cardíaca.
- Insuficiencia renal.
- Inflamación de una vena, coágulos de sangre en las venas y pulmones.
- Problemas de coagulación sanguínea.
- Fallo circulatorio.
- Inflamación de la membrana que rodea el corazón o presencia de líquido alrededor del corazón.
- Infecciones que incluyen infecciones del tracto urinario, gripe, infecciones por virus del herpes, infecciones del oído y celulitis.
- Sangre en las heces o sangrado de las membranas mucosas, por ejemplo, boca, vagina.
- Trastornos cerebrovasculares.
- Parálisis, convulsiones, caídas, trastornos del movimiento, sensibilidad anormal, alterada o reducida (sensación, oído, gusto, olfato), alteración de la atención, temblores, espasmos.
- Artritis, incluida la inflamación de las articulaciones de los dedos de las manos, los pies y la mandíbula.
- Trastornos que afectan los pulmones, impidiendo que su cuerpo reciba suficiente oxígeno. Algunos de estos incluyen dificultad para respirar, falta de aire, sibilancias incluso sin actividad física, dificultad para respirar superficialmente o necesidad de detenerse, sibilancias.
- Hipo, trastornos del habla.
- Aumento o disminución de la producción de orina (daño renal), dolor al orinar o sangre / proteínas en la orina, retención de líquidos.
- Niveles alterados de conciencia, confusión, deterioro o pérdida de la memoria.
- Hipersensibilidad
- Pérdida de audición, sordera o pitidos en los oídos, molestias en el oído.
- Alteraciones hormonales que pueden afectar a la reabsorción de sales y agua.
- Hiperactividad de la glándula tiroides.
- Incapacidad para producir suficiente insulina o resistencia a los niveles normales de insulina.
- Ojos doloridos o inflamados, ojos excesivamente húmedos, dolor ocular, ojo seco, infecciones oculares, secreción ocular, alteraciones de la visión, hemorragia ocular.
- Ganglios linfáticos agrandados.
- Rigidez de articulaciones o músculos, sensación de pesadez, dolor en la ingle.
- Caída del cabello o textura anormal del cabello.
- Reacciones alérgicas.
- Enrojecimiento o dolor en el lugar de la inyección.
- Dolor en la boca
- Infección o inflamación de la boca, úlceras en la boca, esófago, estómago e intestinos a veces asociadas con dolor o sangrado, alteración de la motilidad intestinal (incluido bloqueo intestinal), malestar abdominal o esofágico, dificultad para tragar, vómitos de sangre.
- Infecciones cutáneas.
- Infecciones bacterianas y virales.
- Infección dental.
- Inflamación del páncreas, obstrucción de las vías biliares.
- Dolor en los genitales, problemas de erección.
- Aumento de peso.
- Sensación de sed.
- Hepatitis.
- Trastornos en el lugar de la inyección o en el lugar del catéter.
- Reacciones o trastornos cutáneos (que pueden ser graves y potencialmente mortales), ulceraciones cutáneas.
- Contusiones, caídas y heridas.
- Inflamación o sangrado de los vasos sanguíneos que pueden manifestarse como pequeños puntos rojos o morados (generalmente en las piernas) que pueden volverse similares a grandes hematomas en la piel o tejidos.
- Quistes benignos.
- Afección cerebral grave y reversible que incluye convulsiones, presión arterial alta, dolor de cabeza, fatiga, confusión, ceguera u otros problemas de visión.
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 personas)
- Problemas cardíacos que incluyen ataque cardíaco, angina de pecho.
- Se ruboriza.
- Decoloración de las venas.
- Inflamación del nervio espinal.
- Problemas de oído, sangrado del oído.
- Actividad reducida de la glándula tiroides.
- Síndrome de Budd-Chiari (los signos clínicos son causados por el bloqueo de las venas del hígado).
- Cambio o función intestinal anormal.
- Hemorragia cerebral (sangrado).
- Decoloración amarilla de los ojos y la piel (ictericia).
- Los signos de una reacción alérgica grave (shock anafiláctico) incluyen dificultad para respirar, dolor en el pecho u opresión en el pecho y / o sensación de mareo / debilidad, picazón intensa en la piel o bultos en la piel, hinchazón de la cara, labios, lengua y / o garganta que pueden causar dificultad para tragar, colapso.
- Trastornos mamarios.
- Secreción vaginal.
- Hinchazón de los genitales.
- Incapacidad para tolerar el consumo de alcohol.
- Pérdida o pérdida de masa corporal.
- Apetito incrementado.
- Fístulas.
- Derrame articular.
- Quiste en la membrana que recubre las articulaciones (quistes sinoviales).
- Fracturas
- Rotura de las fibras musculares que conduce a otras complicaciones.
- Hígado agrandado, hemorragia hepática.
- Cancer de RIÑON.
- Condición de la piel similar a la psoriasis.
- Cáncer de piel.
- Palidez de la piel.
- Aumento de plaquetas o células plasmáticas (un tipo de glóbulo blanco) en la sangre.
- Reacción anormal a la transfusión de sangre.
- Pérdida total o parcial de la visión.
- Disminución de la libido.
- Pérdida de saliva.
- Protuberancia del ojo.
- Fotofobia (sensibilidad excesiva del ojo a la luz).
- Respiración rápida.
- Dolor en el recto.
- Cálculos biliares.
- Hernia.
- Lesiones
- Uñas quebradizas o débiles.
- Depósito anormal de proteínas en órganos vitales.
- Coma.
- Úlceras intestinales.
- Daño a múltiples órganos.
- Muerte.
Si se le administra VELCADE junto con otros medicamentos para tratar el linfoma de células del manto, los efectos secundarios que pueden ocurrir se enumeran a continuación.
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
- Neumonía.
- Pérdida de apetito.
- Sensibilización, entumecimiento, hormigueo o sensación de ardor en la piel, o dolor en manos o pies, debido a daño en los nervios.
- Náuseas y vómitos.
- Diarrea.
- Úlceras en la boca.
- Estreñimiento intestinal
- Dolor muscular, dolor de huesos.
- Caída del cabello o textura anormal del cabello.
- Cansancio, sensación de debilidad.
- Fiebre.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas)
- Infección por herpes zóster (localizada, incluso alrededor de los ojos o diseminada por el cuerpo).
- Infección por virus del herpes.
- Infecciones bacterianas y virales.
- Infecciones respiratorias, bronquitis, tos con flema, enfermedad similar a la gripe.
- Infecciones por hongos.
- Hipersensibilidad (reacción alérgica).
- Incapacidad para producir suficiente insulina o resistencia a los niveles normales de insulina.
- Retención de agua.
- Dificultad o problemas para dormir.
- Pérdida de consciencia.
- Niveles alterados de conciencia, confusión.
- Sintiéndose mareado.
- Aumento de la frecuencia cardíaca, presión arterial alta, sudoración.
- Alteraciones visuales, visión borrosa.
- Insuficiencia cardíaca, ataque cardíaco, dolor de pecho, malestar en el pecho, aumento o disminución de la frecuencia cardíaca.
- Presión arterial alta o baja.
- Caída repentina de la presión arterial al estar de pie que puede provocar desmayos.
- Dificultad para respirar durante la actividad física.
- Tos.
- Hipo.
- Zumbidos en los oídos, molestias en el oído.
- Sangrado intestinal o estomacal.
- Dolor de estómago.
- Dolor de estómago, hinchazón.
- Dificultad para tragar.
- Infección o inflamación del estómago y los intestinos.
- Dolor de estómago.
- Irritación de boca o labios, dolor de garganta.
- Alteración de la función hepática.
- Picazón en la piel.
- Enrojecimiento de la piel.
- Sarpullido.
- Espasmos musculares.
- Infección del tracto urinario.
- Dolor en las extremidades.
- Hinchazón del cuerpo, incluida la hinchazón alrededor de los ojos y otras partes del cuerpo.
- Escalofríos.
- Enrojecimiento y dolor en el lugar de la inyección.
- Sensación de malestar general.
- Pérdida de peso corporal.
- Aumento de peso corporal.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
- Hepatitis.
- Reacción alérgica grave (reacción anafiláctica) cuyos signos pueden incluir dificultad para respirar, dolor u opresión en el pecho y / o sensación de mareo / debilidad, picor intenso o bultos en la piel, hinchazón de la cara, labios, lengua y / o sensación de mareo / debilidad. o garganta que puede causar dificultad para tragar, colapso.
- Trastornos del movimiento, parálisis, contracciones.
- Mareo.
- Pérdida de audición, sordera.
- Trastornos que afectan los pulmones, impidiendo que su cuerpo reciba suficiente oxígeno. Algunos de estos incluyen dificultad para respirar, falta de aire, sibilancias incluso sin actividad física, respiración que se vuelve superficial, difícil o se detiene, sibilancias.
- Coágulos de sangre en los pulmones.
- Decoloración amarilla de los ojos y la piel (ictericia).
Notificación de efectos secundarios
Si experimenta cualquier efecto adverso, consulte a su médico o farmacéutico, incluido cualquier posible efecto adverso no mencionado en este prospecto. También puede notificar los efectos secundarios directamente a través del sistema de notificación nacional que figura en el Apéndice V. Al notificar los efectos secundarios, puede ayudar a proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
Mantenga este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el vial y la caja después de CAD.
Almacenar a una temperatura no superior a 30 ° C. Mantenga el vial en el embalaje exterior para proteger el medicamento de la luz.
La solución reconstituida debe usarse inmediatamente después de la preparación. Si la solución reconstituida no se usa inmediatamente, las condiciones y tiempos de uso son responsabilidad del preparador. Sin embargo, la solución reconstituida es estable durante 8 horas a 25 ° C antes de la administración cuando se almacena en el vial original y / o en una jeringa, con un tiempo máximo de almacenamiento para el medicamento reconstituido que no excede las 8 horas.
VELCADE es para un solo uso. El producto no utilizado y los residuos deben eliminarse de acuerdo con las normativas locales vigentes.
Fecha límite "> Otra información
Qué contiene VELCADE
- El ingrediente activo es bortezomib. Cada vial contiene 3,5 mg de bortezomib (como éster borónico de manitol). Después de la reconstitución, 1 ml de solución inyectable contiene 1 mg de bortezomib.
- Los demás componentes son manitol (E421) y nitrógeno.
Reconstitución para uso intravenoso: Después de la reconstitución, 1 ml de solución para inyección intravenosa contiene 1 mg de bortezomib.
Reconstitución para uso subcutáneo: Después de la reconstitución, 1 ml de solución para inyección subcutánea contiene 2,5 mg de bortezomib.
Descripción del aspecto de VELCADE y contenido del envase
VELCADE polvo para solución inyectable es de color blanco a blanco cremoso.
Cada envase de VELCADE 3,5 mg contiene 1 vial de vidrio de 10 ml con tapón azul, contenido en un blister transparente.
Fecha límite "> Información para profesionales sanitarios
La siguiente información está destinada únicamente a profesionales sanitarios.
RECONSTITUCIÓN PARA INYECCIÓN INTRAVENOSA
Nota: VELCADE es un agente citotóxico. En consecuencia, se debe tener especial cuidado durante la manipulación y preparación. Se recomienda usar guantes y otra ropa protectora, para evitar el contacto con la piel.
DEBIDO A LA AUSENCIA DE CUALQUIER TIPO DE CONSERVANTES, SE DEBEN SEGUIR NORMAS TÉCNICAS ASÉPTICAS DURANTE LA MANIPULACIÓN DE VELCADE.
- Preparación del vial de 3,5 mg: Añada 3,5 ml de solución inyectable estéril de cloruro de sodio 9 mg / ml (0,9%) al vial que contiene el polvo de VELCADE. El polvo liofilizado se disuelve completamente en menos de 2 minutos.
La concentración de la solución obtenida es igual a 1 mg / ml. La solución será transparente e incolora con un pH final entre 4 y 7. No es necesario verificar el pH de la solución.
- La solución debe inspeccionarse visualmente antes de la administración para verificar si hay partículas o cambios de color. En presencia de partículas o cambios de color, la solución no debe usarse y debe desecharse.
Confirme la concentración en el vial para asegurarse de que se administra la dosis correcta por vía intravenosa (1 mg / ml).
- La solución reconstituida no contiene conservantes y debe usarse inmediatamente después de la preparación mientras permanece química y físicamente estable durante 8 horas a 25 ° C en el vial original y / o en una jeringa con un máximo de 8 horas en la jeringa. El tiempo total de conservación del medicamento reconstituido no puede exceder las 8 horas antes de la administración. Si la solución reconstituida no se usa inmediatamente después de la preparación, es responsabilidad del usuario cumplir con las condiciones y tiempos de almacenamiento del producto antes de su uso.
No es necesario proteger de la luz el medicamento reconstituido.
ADMINISTRACIÓN
- Una vez disuelta, extraer la cantidad adecuada de solución reconstituida de acuerdo con la dosis calculada en base a la superficie corporal del paciente.
- Confirme la dosis y la concentración en la jeringa antes de usar (verifique que la jeringa esté marcada para administración intravenosa).
- Inyecte la solución por vía intravenosa como un bolo de 3-5 segundos, a través de un catéter intravenoso periférico o central.
- Enjuague el catéter intravenoso con una solución inyectable estéril de cloruro de sodio 9 mg / ml (0,9%).
VELCADE 3,5 mg polvo para solución inyectable ES PARA USO SUBCUTÁNEA O INTRAVENOSO. No administrar por otras vías. La administración intratecal resultó en muertes.
DISPOSICIÓN
El vial es para un solo uso y la solución restante debe desecharse.
Los medicamentos no utilizados y los desechos de este medicamento deben eliminarse de acuerdo con las regulaciones locales.
La siguiente información está destinada únicamente a profesionales sanitarios:
Solo el vial de 3,5 mg se puede administrar por vía subcutánea como se describe a continuación.
RECONSTITUCIÓN POR INYECCIÓN SUBCUTÁNEA
Nota: VELCADE es un agente citotóxico. En consecuencia, se debe tener especial cuidado durante la manipulación y preparación. Se recomienda usar guantes y otra ropa protectora, para evitar el contacto con la piel.
DEBIDO A LA AUSENCIA DE CUALQUIER TIPO DE CONSERVANTES, SE DEBEN SEGUIR NORMAS TÉCNICAS ASÉPTICAS DURANTE LA MANIPULACIÓN DE VELCADE.
- Preparación del vial de 3,5 mg: Añada 1,4 ml de solución inyectable estéril de cloruro de sodio 9 mg / ml (0,9%) al vial que contiene el polvo de VELCADE. El polvo liofilizado se disuelve completamente en menos de 2 minutos.
La concentración de la solución obtenida es igual a 2,5 mg / ml. La solución será transparente e incolora con un pH final entre 4 y 7. No es necesario verificar el pH de la solución.
- La solución debe inspeccionarse visualmente antes de la administración para verificar si hay partículas o cambios de color. En presencia de partículas o cambios de color, la solución no debe usarse y debe desecharse.
Confirme la concentración en el vial para asegurarse de que se administra por vía subcutánea la dosis correcta (2,5 mg / ml).
- La solución reconstituida no contiene conservantes y debe usarse inmediatamente después de la preparación mientras permanece química y físicamente estable durante 8 horas a 25 ° C en el vial original y / o en una jeringa con un máximo de 8 horas en la jeringa. El tiempo total de conservación del medicamento reconstituido no puede exceder las 8 horas antes de la administración. Si la solución reconstituida no se usa inmediatamente después de la preparación, es responsabilidad del usuario cumplir con las condiciones y tiempos de almacenamiento del producto antes de su uso.
No es necesario proteger el medicamento reconstituido de la luz.
ADMINISTRACIÓN
- Una vez disuelta, extraer la cantidad adecuada de solución reconstituida de acuerdo con la dosis calculada en base a la superficie corporal del paciente.
- Confirme la dosis y la concentración en la jeringa antes de usar (verifique que la jeringa esté marcada para administración subcutánea).
- Inyecte la solución por vía subcutánea, en un ángulo de 45-90 °.
- La solución reconstituida se administra por vía subcutánea en los muslos (derecho o izquierdo) o el abdomen (derecho o izquierdo).
- En administraciones posteriores es necesario cambiar el sitio de inyección en rotación.
- Si se producen reacciones locales en el lugar de la inyección después de la inyección subcutánea de VELCADE, se puede administrar una concentración más baja de VELCADE 3,5 mg en solución (1 mg / ml en lugar de 2,5 mg / ml) o se recomienda cambiar a "inyección intravenosa".
VELCADE 3,5 mg polvo para solución inyectable ES PARA USO SUBCUTÁNEA O INTRAVENOSO. No administrar por otras vías. La administración intratecal resultó en muertes.
DISPOSICIÓN
El vial es para un solo uso y la solución restante debe desecharse.
Los medicamentos no utilizados y los desechos de este medicamento deben eliminarse de acuerdo con las regulaciones locales.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO -
VELCADE 3,5 MG POLVO PARA SOLUCIÓN PARA INYECCIÓN
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA -
Cada vial contiene 3,5 mg de bortezomib (como éster borónico de manitol).
Después de la reconstitución, 1 ml de solución inyectable para uso subcutáneo contiene 2,5 mg de bortezomib.
Después de la reconstitución, 1 ml de solución inyectable para uso intravenoso contiene 1 mg de bortezomib.
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA -
Polvo para solución inyectable.
Polvo blanco a blanco cremoso (compactado o no).
04.0 INFORMACIÓN CLÍNICA -
04.1 Indicaciones terapéuticas -
VELCADE en monoterapia o en combinación con doxorrubicina liposomal pegilada o dexametasona está indicado para el tratamiento de pacientes adultos con mieloma múltiple progresivo que ya han recibido al menos una línea de tratamiento anterior y que ya se han sometido o no son elegibles para un trasplante de células madre. .
VELCADE en combinación con melfalán y prednisona está indicado para el tratamiento de pacientes adultos con mieloma múltiple no tratado previamente que no son elegibles para quimioterapia de dosis alta con trasplante de células madre hematopoyéticas.
VELCADE en combinación con dexametasona o con dexametasona y talidomida está indicado para el tratamiento de inducción de pacientes adultos con mieloma múltiple no tratado previamente que son elegibles para quimioterapia de dosis alta con trasplante de células madre hematopoyéticas.
VELCADE en combinación con rituximab, ciclofosfamida, doxorrubicina y prednisona está indicado para el tratamiento de pacientes adultos con linfoma de células del manto no tratado previamente que no son elegibles para un trasplante de células madre hematopoyéticas.
04.2 Posología y forma de administración -
El tratamiento debe iniciarse y administrarse bajo la supervisión de un médico capacitado y con experiencia en el uso de agentes quimioterapéuticos. VELCADE debe ser reconstituido por un profesional de la salud.
Posología para el tratamiento del mieloma múltiple progresivo (pacientes que han recibido al menos una línea de tratamiento anterior)
Monoterapia
VELCADE 3,5 mg polvo para solución inyectable se administra por vía intravenosa o subcutánea a la dosis recomendada de 1,3 mg / m² de superficie corporal dos veces por semana durante dos semanas los días 1, 4, 8 y 11 en un ciclo de tratamiento de 21 días Este período de 3 semanas se considera un ciclo de tratamiento.
Se recomienda que los pacientes reciban 2 ciclos de VELCADE después de la confirmación de que se logra una respuesta completa.
Se recomienda a los pacientes que responden al tratamiento pero no logran una remisión completa que administren un total de 8 ciclos de VELCADE.
Deben transcurrir al menos 72 horas entre la administración de dos dosis consecutivas de VELCADE.
Ajustes de dosis durante el tratamiento y su reanudación como monoterapia.
El tratamiento con VELCADE se debe interrumpir al inicio de cualquier toxicidad no hematológica de Grado 3 o hematológica de Grado 4, excluida la neuropatía, como se indica a continuación (ver también sección 4.4). 1,3 mg / m² reducido a 1,0 mg / m²; 1,0 mg / m² reducido a 0,7 mg / m²). Cuando los síntomas de toxicidad no se han resuelto, o si reaparecen a una dosis reducida, se debe considerar la suspensión de VELCADE, a menos que los beneficios de la terapia claramente superan los riesgos.
Dolor neuropático y / o neuropatía periférica
Los pacientes que experimenten dolor neuropático relacionado con bortezomib y / o neuropatía periférica deben tratarse de acuerdo con la Tabla 1 (ver sección 4.4).
Los pacientes con neuropatía grave preexistente solo pueden ser tratados con VELCADE después de una "evaluación cuidadosa de riesgo / beneficio".
Tabla 1: Modificaciones de dosis recomendadas * en caso de neuropatía relacionada con la administración de bortezomib
Terapia combinada con doxorrubicina liposomal pegilada
VELCADE 3,5 mg polvo para solución inyectable se administra por vía intravenosa o subcutánea a la dosis recomendada de 1,3 mg / m² de superficie corporal dos veces por semana durante dos semanas los días 1, 4, 8 y 11 en un ciclo de tratamiento de 21 días Este período de 3 semanas se considera un ciclo de tratamiento Deben transcurrir al menos 72 horas entre la administración de dos dosis consecutivas de VELCADE.
La doxorrubicina liposomal pegilada se administra a una dosis de 30 mg / m² el día 4 del ciclo de tratamiento de VELCADE como una infusión intravenosa que dura 1 hora después de la inyección de VELCADE.
Se pueden administrar hasta 8 ciclos de esta terapia combinada hasta que los pacientes muestren progresión y toleren el tratamiento. Los pacientes que logran una respuesta completa pueden continuar el tratamiento durante al menos 2 ciclos después de la primera evidencia de respuesta completa, incluso si esto requiere tratamiento durante más de 8 ciclos. Los pacientes cuyos niveles de paraproteína continúan disminuyendo después de 8 ciclos pueden continuar la terapia siempre que se tolere el tratamiento y continúe mostrando una respuesta.
Para obtener más información sobre la doxorrubicina liposomal pegilada, consulte el Resumen de las características del producto correspondiente.
Combinación con dexametasona
VELCADE 3,5 mg polvo para solución inyectable se administra por vía intravenosa o subcutánea a la dosis recomendada de 1,3 mg / m² de superficie corporal dos veces por semana durante dos semanas los días 1, 4, 8 y 11 en un ciclo de tratamiento de 21 días Este período de 3 semanas se considera un ciclo de tratamiento Deben transcurrir al menos 72 horas entre la administración de dos dosis consecutivas de VELCADE.
La dexametasona se administra por vía oral a una dosis de 20 mg los días 1, 2, 4, 5, 8, 9, 11 y 12 del ciclo de tratamiento con VELCADE.
Los pacientes que logran una respuesta o estabilización de la enfermedad después de 4 ciclos de esta terapia combinada pueden continuar recibiendo la misma combinación hasta por 4 ciclos adicionales.
Para obtener más información sobre la dexametasona, consulte el Resumen de las características del producto correspondiente.
Ajuste de dosis para la terapia combinada en pacientes con mieloma múltiple progresivo
Para ajustar la dosis de VELCADE en terapia combinada, siga las recomendaciones de modificación de dosis descritas en la sección de monoterapia anterior.
Posología para el tratamiento del mieloma múltiple no tratado previamente en pacientes no aptos para quimioterapia de dosis alta con trasplante de células madre hematopoyéticas.
Terapia combinada con melfalán y prednisona.
VELCADE 3,5 mg polvo para solución inyectable se administra por vía intravenosa o subcutánea en combinación con melfalán oral y prednisona oral como se indica en la Tabla 2. Un período de 6 semanas se considera un curso de tratamiento. En los ciclos 1-4, VELCADE se administra dos veces por semana los días 1, 4, 8, 11, 22, 25, 29 y 32. En los ciclos 5-9, VELCADE se administra una vez a la semana los días 1, 8, 22 y 29. Deben transcurrir al menos 72 horas entre la administración de dos dosis consecutivas de VELCADE.
Tanto el melfalán como la prednisona deben administrarse por vía oral los días 1, 2, 3 y 4 de la primera semana de cada ciclo de tratamiento con VELCADE. Se administran 9 ciclos de tratamiento de esta terapia de combinación.
Tabla 2: Lista de posología recomendada de VELCADE en combinación con melfalán y prednisona
Ajustes de dosis durante el tratamiento y su reanudación en combinación con melfalán y prednisona.
Antes de comenzar un nuevo curso de terapia:
• El recuento de plaquetas debe ser ≥ 70 x 109 / L y el recuento absoluto de neutrófilos (RAN) ≥ 1,0 x 109 / L
• Las toxicidades no hematológicas deben haber disminuido a Grado 1 o al inicio.
Tabla 3: Cambios en la posología durante los ciclos posteriores de terapia con VELCADE en combinación con melfalán y prednisona
Para obtener más información sobre el melfalán y la prednisona, consulte sus respectivos Resúmenes de las características del producto.
Posología para el tratamiento del mieloma múltiple no tratado previamente en pacientes aptos para un trasplante de células madre hematopoyéticas (terapia de inducción).
Terapia combinada con dexametasona
VELCADE 3,5 mg polvo para solución inyectable se administra por vía intravenosa o subcutánea a la dosis recomendada de 1,3 mg / m² de superficie corporal dos veces por semana durante dos semanas los días 1, 4, 8 y 11 en un ciclo de tratamiento de 21 días Este período de 3 semanas se considera un ciclo de tratamiento Deben transcurrir al menos 72 horas entre la administración de dos dosis consecutivas de VELCADE.
La dexametasona se administra por vía oral a una dosis de 40 mg los días 1, 2, 3, 4, 8, 9, 10 y 11 del ciclo de tratamiento con VELCADE.
Se administran cuatro ciclos de tratamiento de esta terapia de combinación.
Terapia combinada con talidomida y dexametasona.
VELCADE 3,5 mg polvo para solución inyectable se administra por vía intravenosa o subcutánea a la dosis recomendada de 1,3 mg / m² de superficie corporal dos veces por semana durante dos semanas los días 1, 4, 8 y 11 en un ciclo de tratamiento de 28 días Este período de 4 semanas se considera un ciclo de tratamiento.
Deben transcurrir al menos 72 horas entre la administración de dos dosis consecutivas de VELCADE.
La dexametasona se administra por vía oral a una dosis de 40 mg los días 1, 2, 3, 4, 8, 9, 10 y 11 del ciclo de tratamiento con VELCADE.
La talidomida se administra por vía oral a una dosis diaria de 50 mg los días 1-14; si se tolera, la dosis se aumenta a 100 mg los días 15 a 28 y posteriormente se puede aumentar a 200 mg por día a partir del ciclo 2 (ver Tabla 4).
Se administran cuatro ciclos de tratamiento de esta terapia de combinación.
Para los pacientes que logran al menos una respuesta parcial, se recomiendan 2 ciclos de tratamiento adicionales.
Tabla 4: Posología de la terapia combinada con VELCADE para el tratamiento del mieloma múltiple no tratado previamente en pacientes elegibles para un trasplante de células madre hematopoyéticas.
Ajuste de dosis para candidatos a trasplante
Para el ajuste de la dosis de VELCADE para la neuropatía, consulte la Tabla 1.
Además, cuando VELCADE se administra en combinación con otros agentes quimioterapéuticos, se debe considerar la reducción de la dosis apropiada de estos medicamentos en caso de toxicidad de acuerdo con las recomendaciones del Resumen de las Características del Producto correspondiente.
Posología para pacientes con linfoma de células del manto (MCL) no tratado previamente
Terapia combinada con rituximab, ciclofosfamida, doxorrubicina y prednisona (VcR-CAP)
VELCADE 3,5 mg polvo para solución inyectable se administra por vía intravenosa o subcutánea a la dosis recomendada de 1,3 mg / m² de superficie corporal dos veces por semana durante dos semanas los días 1, 4, 8 y 11, seguido de un período de descanso de 10 días los días 12 a 21. Este período de 3 semanas se considera un curso de tratamiento. Deben transcurrir al menos 72 horas entre la administración de dos dosis consecutivas de VELCADE.
Se recomiendan 6 ciclos de tratamiento con esta terapia combinada. Los pacientes con una primera respuesta documentada al ciclo 6 pueden recibir 2 ciclos de tratamiento adicionales.
Los siguientes medicamentos se administran como perfusión intravenosa el día 1 de cada ciclo de tratamiento de 3 semanas con VELCADE: rituximab a una dosis de 375 mg / m², ciclofosfamida a una dosis de 750 mg / m² y doxorrubicina a una dosis de 50 mg. / m².
La prednisona se administra por vía oral a una dosis de 100 mg / m² los días 1, 2, 3, 4 y 5 de cada ciclo de tratamiento con VELCADE.
Ajuste de dosis durante el tratamiento de pacientes con MCL no tratado previamente
Antes de comenzar un nuevo curso de terapia:
• El recuento de plaquetas debe ser ≥ 100.000 células / mcL y el recuento absoluto de neutrófilos (RAN) debe ser ≥ 1.500 células / mcL
• El recuento de plaquetas debe ser ≥ 75.000 células / mcL en pacientes con infiltración de médula ósea o secuestro esplénico
• La hemoglobina debe ser ≥ 8 g / dL
• Las toxicidades no hematológicas deben reducirse a Grado 1 o al inicio.
El tratamiento con VELCADE debe suspenderse al inicio de cualquier toxicidad no hematológica relacionada con VELCADE de Grado ≥ 3 (excluyendo neuropatía) o toxicidad hematológica de Grado ≥ 3 (ver también sección 4.4). Para ajustar la dosis, ver la Tabla 5 a continuación.
En caso de toxicidad hematológica, se pueden administrar factores de crecimiento de granulocitos de acuerdo con la práctica estándar local. Se debe considerar el uso preventivo de factores de crecimiento de granulocitos en caso de retrasos repetidos en la administración de ciclos de terapia Cuando sea clínicamente apropiado, se debe considerar la transfusión de plaquetas para el tratamiento de la trombocitopenia.
Tabla 5: Ajuste de dosis durante el tratamiento de pacientes con MCL no tratado previamente
Además, cuando VELCADE se administra en combinación con otros agentes quimioterapéuticos, "se debe considerar una reducción adecuada de la dosis de estos medicamentos en caso de toxicidad, de acuerdo con las recomendaciones contenidas en el respectivo Resumen de las Características del Producto".
Poblaciones especiales
Pacientes de edad avanzada
No hay evidencia clínica que sugiera la necesidad de ajustar la dosis en pacientes mayores de 65 años con mieloma múltiple o linfoma de células del manto.
No existen estudios sobre el uso de VELCADE en pacientes de edad avanzada con mieloma múltiple no tratado previamente que sean candidatos a quimioterapia de dosis alta con trasplante de células madre hematopoyéticas.
Por lo tanto, no se pueden hacer recomendaciones de dosis en esta población.
En un estudio en pacientes con linfoma de células del manto no tratados previamente, el 42,9% y el 10,4% de los pacientes expuestos a VELCADE estaban en el rango de 65-74 años y ≥ 75 años de edad, respectivamente. En pacientes ≥ 75 años, ambos regímenes, VELCADE en combinación con rituximab, ciclofosfamida, doxorrubicina y prednisona (VcR-CAP) y rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP), fueron menos tolerados (ver párrafo 4.8). ).
Disfunción hepática
Los pacientes con insuficiencia hepática leve no requieren ajuste de dosis y deben ser tratados con la dosis recomendada.Los pacientes con insuficiencia hepática moderada o grave deben iniciar el tratamiento con VELCADE a una dosis reducida de 0,7 mg / m² por inyección durante el primer ciclo de tratamiento, y puede aumentar la dosis posterior a 1,0 mg / m² o una reducción adicional de la dosis a 0,5 mg / m². debe considerarse en función de la tolerancia del paciente (ver Tabla 6 y secciones 4.4 y 5.2).
Tabla 6: Modificaciones de la dosis inicial recomendada de VELCADE para pacientes con insuficiencia hepática
Disfuncion renal
La farmacocinética de bortezomib no se ve afectada en pacientes con insuficiencia renal leve a moderada (aclaramiento de creatinina [CrCL]> 20 ml / min / 1,73 m²); por lo tanto, no se requieren ajustes de dosis en estos pacientes. Se desconoce si la farmacocinética de bortezomib está alterada en pacientes con insuficiencia renal grave que no están en diálisis (CrCL
Población pediátrica
No se ha establecido la seguridad y eficacia de VELCADE en pacientes menores de 18 años (ver secciones 5.1 y 5.2) No hay datos disponibles.
Método de administración
VELCADE 3,5 mg polvo para solución inyectable es para administración intravenosa o subcutánea.
VELCADE 1 mg polvo para solución inyectable es solo para administración intravenosa.
VELCADE no debe administrarse por otras vías. La administración intratecal provocó la muerte.
Inyección intravenosa
La solución reconstituida de VELCADE 3,5 mg se administra por vía intravenosa como un bolo de 3-5 segundos, a través de un catéter intravenoso periférico o central, seguido de lavado con cloruro de sodio 9 mg / ml (0, 9%). Debe haber al menos 72 horas entre dos dosis consecutivas de VELCADE.
Inyección subcutánea
La solución reconstituida de VELCADE 3,5 mg se administra por vía subcutánea en los muslos (derecho o izquierdo) o el abdomen (derecho o izquierdo) La solución se inyecta por vía subcutánea en un ángulo de 45-90 °.
Los lugares de inyección deben cambiarse en rotación para las inyecciones posteriores.
Si se producen reacciones en el lugar de la inyección después de la administración subcutánea de VELCADE, se puede administrar una solución menos concentrada de VELCADE (VELCADE 3,5 mg reconstituido a 1 mg / ml en lugar de 2,5 mg / ml) por vía subcutánea o se recomienda cambiar a la administración intravenosa.
Cuando VELCADE se administra en combinación con otros medicamentos, consulte el Resumen de las características del producto de estos medicamentos para obtener instrucciones sobre la administración.
04.3 Contraindicaciones -
Hipersensibilidad al principio activo, al boro oa alguno de los excipientes incluidos en la sección 6.1.
Enfermedad pulmonar infiltrativa aguda difusa y pericardiopatía.
Cuando VELCADE se administra en combinación con otros medicamentos, consulte el Resumen de las características del producto correspondiente para conocer las contraindicaciones adicionales.
04.4 Advertencias especiales y precauciones de uso apropiadas -
Cuando VELCADE se administra en combinación con otros medicamentos, se debe consultar el Resumen de las Características del Producto correspondiente antes de iniciar el tratamiento con VELCADE. Se debe prestar especial atención a las pruebas de embarazo y las normas de prevención del embarazo cuando se administra talidomida (ver sección 4.6).
Administración intratecal
Ha habido casos de muerte tras la administración inadvertida de VELCADE por vía intratecal. VELCADE 1 mg polvo para solución inyectable está destinado únicamente para uso intravenoso, mientras que VELCADE 3,5 mg polvo para solución inyectable está destinado a uso intravenoso o subcutáneo. VELCADE no debe administrarse por vía intratecal.
Toxicidad gastrointestinal
Los efectos tóxicos gastrointestinales, que incluyen náuseas, diarrea, vómitos y estreñimiento, son muy frecuentes durante el tratamiento con VELCADE. Con poca frecuencia se han notificado casos de íleo paralítico (ver sección 4.8). Por lo tanto, los pacientes que experimentan estreñimiento deben ser monitoreados de cerca.
Toxicidad hematológica
El tratamiento con VELCADE se asocia muy a menudo con efectos tóxicos hematológicos (trombocitopenia, neutropenia y anemia). En estudios realizados en pacientes con mieloma múltiple en recaída tratados con VELCADE y en pacientes con MCL no tratado previamente tratados con VELCADE en combinación con rituximab, ciclofosfamida, doxorrubicina y prednisona (VcR-CAP), una de las toxicidades hematológicas más frecuentes fue la trombocitopenia transitoria. Las plaquetas se encontraban en su nivel más bajo el día 11 de cada ciclo de tratamiento con VELCADE y volvieron a los niveles iniciales, por lo general, en el siguiente ciclo. No hubo evidencia de trombocitopenia acumulativa. El nadir medio del valor plaquetario fue aproximadamente el 40% del valor inicial en los estudios de mieloma múltiple con VELCADE utilizado como monoterapia y el 50% en el estudio MCL. En pacientes con mieloma avanzado, la gravedad de la trombocitopenia se relacionó con los valores de plaquetas previos al tratamiento: para los valores de plaquetas en el valor inicial de 75.000 / mcl, solo el 14% de 309 pacientes tenían recuentos de plaquetas ≤ 25.000 / mcl durante el estudio.
En pacientes con MCL (estudio LYM-3002), hubo una mayor incidencia (56,7% versus 5,8%) Trombocitopenia de grado ≥ 3 en el grupo de tratamiento con VELCADE (VcR-CAP) versus el grupo sin VELCADE (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona [R-CHOP]). Los dos grupos de tratamiento fueron similares tanto en la incidencia general de episodios hemorrágicos de todos los grados (6,3% en el grupo VcR-CAP y 5,0% en el grupo R-CHOP) como en el grado 3 y hemorragia mayor (VcR-CAP: 4 pacientes [ 1,7%]; R-CHOP: 3 pacientes [1,2%]) En el grupo VcR-CAP, el 22,5% de los pacientes recibieron una transfusión de plaquetas en comparación con el 2,9% de los pacientes del grupo R-CHOP.
Se han notificado casos de hemorragia gastrointestinal e intracerebral en asociación con el tratamiento con VELCADE. Por lo tanto, se deben controlar los niveles de plaquetas antes de la administración de cada dosis de VELCADE. La terapia con VELCADE debe suspenderse cuando el recuento de plaquetas alcance los valores.
Los recuentos sanguíneos completos, con recuentos diferenciales e incluyendo recuentos de plaquetas, deben controlarse con frecuencia durante el tratamiento con VELCADE. Cuando sea clínicamente apropiado, se debe considerar la transfusión de plaquetas (ver sección 4.2).
En pacientes con MCL sin evidencia de neutropenia acumulativa, se observó neutropenia transitoria reversible entre ciclos de tratamiento. Los neutrófilos se encontraban en su nivel más bajo el día 11 de cada ciclo de tratamiento con VELCADE y, por lo general, volvían a los valores iniciales en el siguiente ciclo. En el estudio LYM-3002, se utilizó un factor de crecimiento de apoyo en el 78% de los pacientes del grupo de VcR-CAP y en el 61% de los pacientes del grupo de R-CHOP. Dado que los pacientes con neutropenia tienen un mayor riesgo de infecciones, se les debe vigilar para detectar signos y síntomas de infección y tratarlos de inmediato. Se pueden administrar factores de crecimiento de granulocitos para tratar la toxicidad hematológica de acuerdo con la práctica estándar local. En caso de retrasos repetidos en la administración de ciclos de terapia, se debe considerar el uso preventivo de factores de crecimiento de granulocitos (ver sección 4.2).
Reactivación del virus del herpes zóster
Se recomienda la administración de profilaxis antiviral en pacientes que reciben VELCADE. En el estudio de fase III realizado en pacientes con mieloma múltiple no tratado previamente, la incidencia global de reactivación del herpes zóster fue más común en pacientes tratados con VELCADE + Melfalán + Prednisona que en pacientes tratados con Melfalán + Prednisona (respectivamente 14% versus 4%).
En pacientes con MCL (estudio LYM-3002), la incidencia de infección por herpes zóster fue del 6,7% en el grupo de VcR-CAP y del 1,2% en el grupo de R-CHOP (ver sección 4.8).
Reactivación e infección por el virus de la hepatitis B (VHB)
Cuando se utiliza rituximab en combinación con VELCADE, la detección del VHB siempre debe realizarse antes de iniciar el tratamiento en pacientes con riesgo de infección por VHB. Los portadores de hepatitis B y los pacientes con antecedentes de hepatitis B deben ser monitoreados de cerca para detectar signos clínicos y de laboratorio de infección activa por VHB durante y después del tratamiento con rituximab en combinación con VELCADE. Se debe considerar la profilaxis antiviral. Para obtener más información, consulte el Resumen de las características del producto de rituximab.
Leucoencefalopatía multifocal progresiva (LMP)
Se han notificado casos muy raros de infección por el virus John Cunningham (JC) que provocaron leucoencefalopatía multifocal progresiva y muerte en pacientes tratados con VELCADE, con causalidad desconocida. Los pacientes diagnosticados con leucoencefalopatía multifocal progresiva habían recibido previamente tratamiento inmunosupresor o lo estaban tomando de forma concomitante. La mayoría de los casos de leucoencefalopatía multifocal progresiva se diagnosticaron dentro de los 12 meses posteriores a la administración de la primera dosis de VELCADE. Los pacientes deben ser monitoreados a intervalos regulares para detectar cualquier síntoma o signo neurológico nuevo o que empeore que pueda indicar LMP entre los diagnósticos diferenciales de problemas del sistema nervioso central. Si se sospecha un diagnóstico de leucoencefalopatía multifocal progresiva, se debe derivar a los pacientes a un médico que se especialice en el tratamiento de la leucoencefalopatía multifocal progresiva y se deben implementar las medidas de diagnóstico adecuadas para la leucoencefalopatía multifocal progresiva. En caso de un diagnóstico confirmado de leucoencefalopatía multifocal progresiva, se debe interrumpir el tratamiento con VELCADE.
Neuropatía periférica
El tratamiento con VELCADE se asocia con mayor frecuencia con la aparición de neuropatía periférica, principalmente sensorial, sin embargo, se han notificado casos de neuropatía motora grave con o sin neuropatía sensorial periférica.
La incidencia de neuropatía periférica aumenta al principio del tratamiento y alcanza su punto máximo en el ciclo 5.
Los pacientes deben ser monitoreados de cerca para detectar síntomas de neuropatía como sensación de ardor, hiperestesia, hipoestesia, parestesia, malestar, dolor neuropático o debilidad.
En el estudio clínico de fase III que comparó VELCADE administrado por vía intravenosa con la vía subcutánea, la incidencia de episodios de neuropatía periférica de grado? 2 fue del 24% en el grupo de administración subcutánea y del 41% en el grupo de inyección intravenosa (p = 0,0124). Grado? 3 periférico La neuropatía se produjo en el 6% de los pacientes en el grupo de tratamiento subcutáneo en comparación con el 16% en el grupo de tratamiento intravenoso (p = 0,0264). Todos los grados de neuropatía periférica con VELCADE administrado por vía intravenosa fueron menores en estudios previos en los que VELCADE se administró por vía intravenosa que en el estudio MMY. -3021.
Se recomienda una evaluación neurológica en pacientes con aparición o empeoramiento de neuropatía periférica, para quienes puede ser necesario un cambio de dosis o régimen o un cambio en la vía de administración por vía subcutánea (ver sección 4.2). La neuropatía se manejó con terapias de apoyo o de otro tipo.
Se debe considerar la monitorización temprana y regular de los síntomas de neuropatía relacionada con el tratamiento con una evaluación neurológica en pacientes que reciben VELCADE en combinación con medicamentos que se sabe que están asociados con neuropatía (por ejemplo, talidomida) y se debe considerar la reducción de la dosis o la interrupción del tratamiento. .
Además de la neuropatía periférica, la neuropatía autonómica puede contribuir a la aparición de algunas reacciones adversas, como la hipotensión postural y el estreñimiento por íleo severo.Aún se dispone de información limitada sobre la neuropatía autonómica y su contribución a estos efectos secundarios.
Convulsiones
Con poca frecuencia se han notificado convulsiones en pacientes sin antecedentes de convulsiones o epilepsia. Se requiere especial cuidado al tratar a pacientes con riesgo de convulsiones.
Hipotensión
El tratamiento con VELCADE se asocia comúnmente con hipotensión ortostática / postural. La mayoría de las reacciones adversas son de gravedad leve a moderada y se han observado durante el tratamiento. Los pacientes que experimentaron hipotensión ortostática con VELCADE (inyectado por vía intravenosa) no habían tenido episodios previos de hipotensión ortostática antes del tratamiento. En la mayoría de los pacientes se requirió terapia para el tratamiento de la hipotensión ortostática. Una minoría de pacientes con hipotensión ortostática experimentó episodios de síncope. La hipotensión ortostática / postural no se relacionó de forma aguda con la infusión en bolo de VELCADE.
Se desconoce el mecanismo de este evento, aunque un componente puede estar determinado por neuropatía autónoma. La neuropatía autónoma puede estar relacionada con bortezomib o es posible que el fármaco agrave una afección preexistente, como la neuropatía diabética o amiloidótica. Se debe tener la máxima precaución en el tratamiento de pacientes con antecedentes de síncope que estén siendo tratados con medicamentos que se sabe que están relacionados con la hipotensión, o en pacientes que presenten deshidratación como resultado de diarrea o vómitos recurrentes. La hipotensión ortostática / postural puede tratarse con un ajuste de dosis de medicamentos antihipertensivos, rehidratación o administración de mineralocorticosteroides y / o medicamentos simpaticomiméticos Se debe advertir a los pacientes que consulten a su médico en caso de mareos, aturdimiento o episodios breves de desmayo.
Síndrome de encefalopatía posterior reversible (PRES)
Se han notificado casos de SEPR en pacientes que recibieron VELCADE. El PRES es una forma neurológica rara caracterizada por una evolución rápida, a menudo reversible, que puede manifestarse con convulsiones, hipertensión, dolor de cabeza, letargo, confusión, ceguera y otros cambios visuales y neurológicos. El diagnóstico se confirma mediante imágenes radiológicas de las estructuras cerebrales, preferiblemente obtenidas con Resonancia Magnética Nuclear (MRI). En pacientes que desarrollen PRES, se debe suspender la terapia con VELCADE.
Insuficiencia cardiaca
Durante el tratamiento con bortezomib se ha observado un inicio o agravamiento agudo de la insuficiencia cardíaca congestiva y / o el desarrollo de una fracción de eyección ventricular izquierda disminuida. La retención de líquidos puede ser un factor predisponente para los signos y síntomas de insuficiencia cardíaca. Pacientes con insuficiencia cardíaca o con factores de riesgo para la insuficiencia cardíaca se debe controlar cuidadosamente.
Investigaciones electrocardiográficas
En estudios clínicos se han observado casos aislados de prolongación del intervalo QT, cuya causalidad no se ha establecido.
Alteraciones pulmonares
Se han notificado casos raros de enfermedad pulmonar infiltrativa difusa aguda de etiología desconocida, como neumonía, neumonía intersticial, infiltración pulmonar y síndrome de dificultad respiratoria aguda (SDRA), en pacientes que recibieron VELCADE (ver sección 4.8). Algunos de estos episodios han resultado fatales. Se recomienda una radiografía de tórax antes del tratamiento como referencia inicial para posibles cambios pulmonares después del tratamiento.
En caso de aparición o empeoramiento de los síntomas pulmonares (p. Ej. Tos, disnea), se debe realizar una evaluación diagnóstica inmediata del paciente y el consiguiente tratamiento adecuado. Se debe considerar el balance riesgo / beneficio antes de continuar el tratamiento con VELCADE.
Durante un estudio clínico, dos de cada dos pacientes que recibieron citarabina en dosis altas (2 g / m² por día) como infusión continua de 24 horas en combinación con daunorrubicina y VELCADE para el tratamiento de la leucemia mieloide aguda recidivante murieron debido a SDRA. la fase inicial de la terapia, el estudio se detuvo. Por lo tanto, no se recomienda este régimen de terapia de combinación específica con dosis altas de citarabina (2 g / m² por día) en perfusión continua de 24 horas.
Insuficiencia renal
Las complicaciones renales son frecuentes en pacientes con mieloma múltiple. Los pacientes con insuficiencia renal deben ser controlados cuidadosamente (ver secciones 4.2 y 5.2).
Función hepática alterada
Bortezomib es metabolizado por enzimas hepáticas. En pacientes con insuficiencia hepática moderada o grave, la exposición a bortezomib aumenta; estos pacientes deben ser tratados con una dosis reducida de VELCADE y deben ser monitoreados cuidadosamente para detectar cualquier aparición de toxicidad (ver secciones 4.2 y 5.2).
Reacciones hepáticas
Se han notificado casos raros de insuficiencia hepática en pacientes que reciben VELCADE y tratamientos farmacológicos concomitantes y con enfermedad subyacente grave.Se han notificado otras reacciones hepáticas como aumento de las enzimas hepáticas, hiperbilirrubinemia y hepatitis. Estos cambios pueden ser reversibles tras la interrupción del tratamiento con bortezomib (ver sección 4.8).
Síndrome de lisis tumoral
Dado que bortezomib es una sustancia citotóxica y, por lo tanto, es capaz de destruir rápidamente las células plasmáticas malignas y las células del MCL, se pueden observar complicaciones del síndrome de lisis tumoral. Los pacientes con riesgo de desarrollar síndrome de lisis tumoral son aquellos que han mostrado una alta carga tumoral antes de iniciar el tratamiento, estos pacientes deben ser monitoreados cuidadosamente y tomar precauciones.
Administración concomitante de otros fármacos
Los pacientes en tratamiento concomitante con bortezomib e inhibidores potentes de CYP3A4 deben ser controlados cuidadosamente. Se debe tener especial cuidado cuando se coadministra bortezomib y sustratos de CYP3A4 o CYP2C19 (ver sección 4.5).
La función hepática normal debe confirmarse en pacientes que reciben hipoglucemiantes orales y deben tratarse con precaución (ver sección 4.5).
Reacciones potencialmente mediadas por complejos inmunes
Se han notificado con poca frecuencia posibles reacciones relacionadas con complejos inmunitarios, como enfermedad del suero, poliartritis con erupción cutánea y glomerulonefritis proliferativa. Se debe interrumpir la administración de bortezomib en caso de eventos graves.
04.5 Interacciones con otros medicamentos y otras formas de interacción -
Educación in vitro indican que bortezomib es un inhibidor débil de las isoenzimas del citocromo P450 (CYP) 1A2, 2C9, 2C19, 2D6 y 3A4. Dada la contribución limitada (7%) de la isoenzima CYP2D6 al metabolismo de bortezomib, se cree que este fenotipo de metabolismo bajo no afecta la disponibilidad general de bortezomib.
Un estudio de interacción fármaco-fármaco, basado en datos de 12 pacientes, para investigar el efecto del ketoconazol, un potente inhibidor del CYP3A4, sobre la farmacocinética de bortezomib (inyectado por vía intravenosa) mostró un aumento medio del AUC. 35% de bortezomib (IC del 90% [ 1.032-1.772)] Por lo tanto, los pacientes que reciben tratamiento concomitante con bortezomib e inhibidores potentes del CYP3A4 (por ejemplo, ketoconazol, ritonavir) deben ser monitoreados de cerca.
En un estudio de interacción fármaco-fármaco, basado en datos de 17 pacientes, para investigar el efecto del omeprazol, un potente inhibidor de CYP2C19, sobre la farmacocinética de bortezomib (inyectado por vía intravenosa) no hubo evidencia de un efecto significativo sobre la farmacocinética de bortezomib. .
Un estudio clínico de interacción fármaco-fármaco, basado en datos de 6 pacientes, para investigar el efecto de la rifampicina, un potente inductor del CYP3A4, sobre la farmacocinética de bortezomib (inyectado por vía intravenosa) mostró una reducción media del AUC. 45% de bortezomib. Por tanto, concomitante. No se recomienda el uso de bortezomib con inductores potentes de CYP3A4 (p. ej., rifampicina, carbamazepina, fenitoína, fenobarbital y hierba de San Juan) ya que la eficacia puede verse reducida.
En el mismo estudio clínico de interacción fármaco-fármaco, con datos de 7 pacientes, para verificar el efecto de la dexametasona, un inductor débil de CYP3A4, sobre la farmacocinética de bortezomib (inyectado por vía intravenosa), no hubo un efecto significativo sobre la farmacocinética de bortezomib.
Un estudio de interacción fármaco-fármaco, basado en datos de 21 pacientes, para evaluar el efecto de melfalán-prednisona sobre la farmacocinética de bortezomib (inyectado por vía intravenosa), mostró un aumento del AUC de bortezomib en un 17%.
Esto no se consideró clínicamente relevante.
En ensayos clínicos, se notificaron hipoglucemia e hiperglucemia con poca frecuencia en pacientes diabéticos que recibían hipoglucemiantes orales. Los pacientes en tratamiento con antidiabéticos orales que reciben VELCADE pueden requerir un control cuidadoso de la glucosa en sangre y un ajuste de la dosis de los fármacos antidiabéticos.
04.6 Embarazo y lactancia -
Anticoncepción en hombres y mujeres
Los hombres y mujeres en edad fértil deben utilizar medidas anticonceptivas adecuadas durante la administración y durante los 3 meses siguientes al tratamiento.
El embarazo
No se dispone de datos clínicos sobre la exposición a bortezomib durante el embarazo El potencial teratogénico de bortezomib no se ha investigado completamente.
En estudios preclínicos, la administración de bortezomib a las dosis máximas toleradas por la madre no mostró ningún efecto sobre el desarrollo embriofetal en ratas y conejos. No se han realizado estudios en animales para determinar ningún efecto sobre el parto y el desarrollo posnatal (ver sección 5.3). VELCADE no debe usarse durante el embarazo a menos que la condición clínica de la paciente requiera su uso.
Se debe informar a la paciente de los riesgos potenciales para el feto si VELCADE se administra durante el embarazo o si la paciente queda embarazada durante el tratamiento.
La talidomida es un potente teratógeno en humanos e induce defectos congénitos graves y potencialmente mortales. La talidomida está contraindicada durante el embarazo y en mujeres en edad fértil, a menos que se cumplan todas las condiciones del programa de prevención del embarazo con talidomida. Las pacientes que reciben VELCADE en combinación con talidomida deben cumplir con el Programa de Prevención de Embarazo con talidomida. Consulte el Resumen de las Características del Producto de talidomida para obtener información adicional.
Hora de la comida
Se desconoce si bortezomib se excreta en la leche materna. Debido al potencial de reacciones adversas graves de VELCADE en bebés amamantados, se debe interrumpir la lactancia durante el tratamiento con VELCADE.
Fertilidad
No se han realizado estudios de fertilidad con VELCADE (ver sección 5.3).
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas.
VELCADE puede afectar moderadamente a la capacidad para conducir o utilizar máquinas.
VELCADE puede asociarse muy a menudo con fatiga, comúnmente con mareos, con poca frecuencia con síncope, comúnmente con hipotensión ortostática / postural o visión borrosa. Los pacientes deben tener extrema precaución al conducir vehículos o utilizar máquinas (ver sección 4.8).
05.0 PROPIEDADES FARMACOLÓGICAS -
05.1 "Propiedades farmacodinámicas -
Grupo farmacoterapéutico: fármacos antineoplásicos, otros fármacos antineoplásicos.
Código ATC: L01XX32.
Mecanismo de acción
Bortezomib es un inhibidor del proteasoma. Está específicamente indicado para inhibir la actividad similar a la quimotripsina del proteasoma 26S en células de mamíferos. El proteasoma 26S es un gran complejo polipeptídico, responsable de la degradación de proteínas ubiquinadas. La ruta metabólica ubiquitina-proteasoma juega un papel esencial en el control del recambio. de proteínas específicas, manteniendo así la homeostasis en las células. La inhibición del proteasoma 26S previene esta proteólisis dirigida y afecta la transmisión de señales dentro de la célula, lo que resulta en la muerte de las células cancerosas.
Bortezomib es muy selectivo para el proteasoma. A concentraciones de 10 mcM, bortezomib no inhibe ninguno de los numerosos receptores y proteasas evaluados y es más de 1.500 veces más selectivo para el proteasoma que la segunda enzima diana. Se evaluó la cinética de inhibición del proteosoma. in vitro y bortezomib se disocia del proteasoma con una semivida de 20 minutos, lo que demuestra que la inhibición de bortezomib es reversible.
La inhibición del proteasoma mediada por bortezomib tiene numerosos efectos sobre las células cancerosas, que incluyen, entre otros, la alteración de las proteínas reguladoras que controlan la progresión del ciclo celular y la "activación del factor nuclear kB (NF-kB). L" inhibición de la el proteasoma conduce a la detención del ciclo celular y la apoptosis.
NF-kB es un factor de transcripción cuya activación se requiere en muchas etapas de la carcinogénesis, incluido el crecimiento y la supervivencia celular, la angiogénesis, la interacción celular y la metástasis. En el mieloma, bortezomib afecta la capacidad de las células de mieloma para interactuar con el microambiente de la médula ósea.
Los ensayos han demostrado que bortezomib es citotóxico para numerosos tipos de células cancerosas y que estas células son mucho más sensibles a los efectos proapoptóticos de la inhibición del proteasoma que las normales. Bortezomib reduce el crecimiento tumoral. en vivo en muchos modelos preclínicos de cáncer, incluido el mieloma múltiple.
Datos in vitro, ex vivo y en modelos animales sugieren que bortezomib aumenta la diferenciación y actividad osteoblástica e inhibe la función osteoclástica Estos efectos se han observado en pacientes con mieloma múltiple con enfermedad osteolítica avanzada y tratados con bortezomib.
Eficacia clínica en mieloma múltiple no tratado previamente
Se realizó un estudio clínico internacional, aleatorizado (1: 1), abierto, prospectivo de fase III (MMY-3002 VISTA) en 682 pacientes para evaluar si VELCADE (Vc) (1,3 mg / m² inyectado por vía intravenosa) en combinación con melfalán ( M) (9 mg / m²) y prednisona (P) (60 mg / m²) mejoraron el tiempo de progresión (TTP) en comparación con melfalán (9 mg / m²) y prednisona (60 mg / m²) en pacientes con mieloma múltiple no tratado previamente . El tratamiento se administró durante hasta 9 ciclos (aproximadamente 54 semanas) y se interrumpió temprano en caso de progresión de la enfermedad o toxicidad inaceptable. En el estudio, la mediana de edad de los pacientes fue de 71 años, el 50% eran hombres, el 88% eran caucásicos y la mediana de la puntuación del estado funcional de Karnofsky de los pacientes fue de 80. Los pacientes tenían IgG / IgA / mieloma de cadenas ligeras en un 63% / 25 % / 8% de los casos, una mediana de hemoglobina de 105 g / ly una mediana del recuento de plaquetas de 221,5 x 109 / l. En los dos grupos, el porcentaje de pacientes que tenían un aclaramiento de creatinina ≤ 30 fue similar. Ml / min ( 3% en cada brazo).
En el momento deanálisis intermedio planificado, se había alcanzado el criterio de valoración principal, el tiempo hasta la progresión, y a los pacientes del grupo M + P se les ofreció el tratamiento Vc + M + P. La mediana de seguimiento fue de 16,3 meses. Después de una mediana de seguimiento La cifra de supervivencia final se actualizó durante 60,1 meses. Se observó un beneficio de supervivencia estadísticamente significativo a favor del grupo de tratamiento Vc + M + P (HR = 0,695, p = 0,00043) a pesar de las terapias posteriores que incluían regímenes basados en VELCADE. Mediana de supervivencia en el tratamiento Vc + M + P grupo fue de 56,4 meses en comparación con 43,1 meses en el grupo de tratamiento M + P. Los resultados de eficacia se muestran en la Tabla 11.
Tabla 11: Resultados de eficacia después de la actualización final de los datos de supervivencia de VISTA
Pacientes candidatos a trasplante de células madre
Se realizaron dos ensayos clínicos multicéntricos de fase III, aleatorizados y abiertos (IFM-2005-01, MMY-3010) para demostrar la seguridad y eficacia de VELCADE en combinación doble y triple con otros agentes quimioterapéuticos como terapia de inducción antes del trasplante de células madre en pacientes sin tratamiento previo para el mieloma múltiple.
En el estudio IFM-2005-01, VELCADE en combinación con dexametasona [VcDx, n = 240] se comparó con vincristina-doxorrubicina-dexametasona [VDDx, n = 242]. Los pacientes del grupo VcDx recibieron cuatro ciclos de 21 días, cada uno de los cuales consistió en VELCADE (1,3 mg / m² administrados por vía intravenosa dos veces a la semana los días 1, 4, 8 y 11) y dexametasona oral (40 mg / m2). los días 1 a 4 y los días 9 a 12, en los Ciclos 1 y 2, y los días 1 a 4 en los Ciclos 3 y 4).
Ciento noventa y ocho pacientes (82%) y 208 pacientes (87%) en los grupos VDDx y VcDx, respectivamente, se habían sometido a un autotrasplante de células madre; la mayoría de los pacientes se sometieron a un solo trasplante. Los datos demográficos de los pacientes y las características iniciales de la enfermedad fueron similares entre los dos grupos de tratamiento. En el estudio, la edad media de los pacientes fue de 57 años, el 55% eran hombres y el 48% de los pacientes tenían un riesgo citogenético alto. La duración media del tratamiento fue de 13 semanas para el grupo VDDx y de 11 semanas para el grupo VDDx. VcDx La mediana del número de ciclos recibidos por ambos grupos fue de 4 ciclos.
El criterio de valoración principal de eficacia del estudio fue la tasa de respuesta posterior a la inducción (CR + nCR). Se observó una diferencia estadísticamente significativa en CR + nCR a favor del grupo VELCADE en combinación con dexametasona. Los criterios de valoración secundarios de eficacia incluyeron tasas de respuesta (CR + nCR, CR + nCR + VGPR + PR) postrasplante, supervivencia libre de progresión y supervivencia global Los principales resultados de eficacia se presentan en la Tabla 12.
Tabla 12: Resultados de eficacia en el estudio IFM-2005-01
En el estudio MMY-3010, VELCADE en combinación con talidomida y dexametasona [VcTDx, n = 130] se comparó con talidomida-dexametasona [TDx, n = 127]. Los pacientes del grupo VcTDx recibieron seis ciclos de 4 semanas, cada uno de los cuales consistió en VELCADE (1,3 mg / m² administrados dos veces por semana los días 1, 4, 8 y 11, seguido de un período de descanso de 17 días 12 a 28), dexametasona (40 mg administrados por vía oral los días 1 a 4 y los días 8 a 11), y talidomida (50 mg diarios administrados por vía oral los días 1-14, con dosis aumentadas hasta 100 mg los días 15-28 y posteriormente a 200 mg por día). día).
Ciento cinco pacientes (81%) y 78 pacientes (61%) en los grupos VcTDx y TDx, respectivamente. se habían sometido a un único trasplante autólogo de células madre. Los datos demográficos de los pacientes y las características iniciales de la enfermedad fueron similares entre los dos grupos de tratamiento. Los pacientes de los grupos VcTDx y TDx, respectivamente, tenían una mediana de edad de 57 y 56 años, el 99% y el 98% de los pacientes eran caucásicos; El 58% y el 54% eran hombres. En el grupo de VcTDx, el 12% de los pacientes se clasificaron citogenéticamente como de alto riesgo en comparación con el 16% de los pacientes del grupo de TDx. La duración media del tratamiento fue de 24,0 semanas y la mediana del número de ciclos de tratamiento recibidos fue de 6,0 y fue constante en todos los grupos de tratamiento.
Los criterios de valoración principales de eficacia del estudio fueron las tasas de respuesta post-inducción y post-trasplante (RC + nCR). Se observó una diferencia estadísticamente significativa en CR + nCR a favor del grupo de tratamiento con VELCADE en combinación con dexametasona y talidomida. Los criterios de valoración secundarios de la eficacia incluyeron la supervivencia libre de progresión y la supervivencia global. Los principales resultados de eficacia se presentan en la Tabla 13.
Tabla 13: Resultados de eficacia del estudio MMY-3010
Eficacia clínica en pacientes con mieloma múltiple en recaída o refractario
Los perfiles de seguridad y eficacia de VELCADE (inyectado por vía intravenosa) se evaluaron en dos estudios a la dosis recomendada de 1,3 mg / m²: un estudio aleatorizado y controlado de dexametasona (Dex) fase III (APEX) realizado en 669 pacientes con mieloma múltiple recidivante y refractario. , que se han sometido de 1 a 3 líneas de tratamiento previas y un estudio de fase II de un solo brazo, realizado en 202 pacientes con mieloma múltiple recidivante y refractario, que se han sometido al menos a dos líneas de tratamiento previas con progresión de la enfermedad después de la última terapia.
En el estudio de Fase III, en todos los pacientes, incluidos los que habían recibido solo una línea previa de terapia, el tratamiento con VELCADE resultó en un alargamiento significativo del tiempo hasta la progresión, una prolongación significativa de la supervivencia y un aumento significativo en la tasa de respuesta. en comparación con el tratamiento con dexametasona (ver Tabla 14).
Según los datos que surgen del "análisis intermedio Preplanificado, el Comité de Seguimiento recomendó la interrupción del tratamiento con dexametasona a favor del tratamiento con VELCADE para todos los pacientes aleatorizados al tratamiento con dexametasona, independientemente del estado de la enfermedad. Debido a este cruce temprano, la duración media del seguimiento de pacientes vivos fue de 8,3 meses. En el grupo de tratamiento con VELCADE, la supervivencia general fue más prolongada y la tasa de respuesta fue mayor tanto en los pacientes que fueron refractarios a la última terapia como en los que no.
De los 669 pacientes incluidos, 245 (37%) tenían 65 años de edad o más. Los parámetros de respuesta y TTP fueron significativamente mejores para VELCADE independientemente de la edad.Todos los parámetros de eficacia (tiempo hasta la progresión, supervivencia general y tasa de respuesta) mejoraron significativamente en el brazo de VELCADE, independientemente de los niveles de b2-microglobulina al inicio.
En la población refractaria del estudio Fase II, las respuestas fueron evaluadas por un comité independiente y los criterios de respuesta aplicados son los establecidos. por el Grupo Europeo de Trasplantes de Médula Ósea. La supervivencia global media de todos los pacientes incluidos en el estudio fue de 17 meses (rango de estado funcional, del estado de deleción del cromosoma 13 o del número o tipo de terapias anteriores. La tasa de respuesta de los pacientes que ya se sometieron a 2-3 o más que 7 líneas de tratamiento fue 32% (10/32) y 31% (21/67), respectivamente.
Tabla 14: Resumen de los resultados de eficacia de los estudios de fase III (APEX) y II
En el estudio de fase II, los pacientes que no lograron una respuesta óptima a la monoterapia con VELCADE fueron tratados con dosis altas de dexametasona y VELCADE. El protocolo permitió a los pacientes que lograron una respuesta menos que óptima a la monoterapia con VELCADE recibir dexametasona.
Un total de 74 pacientes evaluables fueron tratados con dexametasona y VELCADE. El tratamiento combinado permitió obtener una respuesta o una mejora de la respuesta [IM 11% o RP 7%] en el 18% de los pacientes.
Eficacia clínica en pacientes con mieloma múltiple en recaída / refractario con la administración subcutánea de VELCADE
Un ensayo clínico de fase III aleatorizado, abierto y de no inferioridad comparó la eficacia y seguridad de la administración subcutánea de VELCADE frente a la administración intravenosa. Este estudio incluyó a 222 pacientes con mieloma múltiple en recaída / refractario, aleatorizados en una proporción de 2: 1 para recibir 1,3 mg / m² de VELCADE por vía subcutánea o intravenosa durante 8 ciclos. Para aquellos pacientes que no lograron una respuesta óptima a VELCADE solo (menos de Respuesta Completa [CR]) después de 4 ciclos, se les permitió recibir 20 mg de dexametasona en el día de la administración de VELCADE y al día siguiente. Pacientes con neuropatía periférica de grado ≥ 2 o recuentos de plaquetas basales
Este estudio cumplió con el objetivo principal de no inferioridad evaluada en la tasa de respuesta (RC + PR) después de 4 ciclos de VELCADE en monoterapia tanto por vía subcutánea como intravenosa, con una tasa de respuesta del 42% en ambas. Además, variables secundarias de eficacia relacionados con la respuesta y el tiempo hasta el evento mostraron resultados consistentes tanto para la vía subcutánea como para la intravenosa (Tabla 15).
Tabla 15: Resumen de los análisis de eficacia que comparan las administraciones subcutánea e intravenosa de VELCADE
Tratamiento con VELCADE en combinación con doxorrubicina liposomal pegilada (estudio DOXIL-MMY-3001)
Se llevó a cabo un estudio multicéntrico de fase III, abierto, aleatorizado y de grupos paralelos en 646 pacientes que comparó la seguridad y eficacia de VELCADE más doxorrubicina liposomal pegilada frente a VELCADE en monoterapia en pacientes con mieloma múltiple que habían recibido al menos 1 tratamiento anteriormente y que no lo habían hecho. mostró progresión de la enfermedad durante la terapia con antraciclinas. El criterio principal de valoración de la eficacia fue el tiempo hasta la progresión (TTP), mientras que los criterios de valoración secundarios de la eficacia fueron la supervivencia global (SG) y la tasa de respuesta global (ORR: (respuesta completa + respuesta parcial) utilizando los criterios del "Grupo europeo de sangre y médula ósea Trasplante (EBMT).
Los resultados de la "análisis intermedio definido por el protocolo (basado en 249 eventos de TTP) llevó a la terminación anticipada del estudio por eficacia. Este análisis intermedio mostró una reducción del riesgo del 45% de TTP (IC del 95%; 29-57%, p
El análisis final para la supervivencia general (SG) realizado después de una mediana de seguimiento de 8,6 años no mostró diferencias significativas en la SG entre los dos grupos de tratamiento. La mediana de SG fue de 30,8 meses (IC del 95%; 25,2-36,5 meses) para los pacientes tratados con VELCADE. monoterapia y 33,0 meses (IC del 95%; 28,9-37,1 meses) para pacientes en terapia combinada con VELCADE y doxorrubicina liposomal pegilada.
Tratamiento con VELCADE en combinación con dexametasona
En ausencia de una comparación directa entre VELCADE y VELCADE en combinación con dexametasona en pacientes con mieloma múltiple progresivo, se realizó un "análisis estadístico pareado" para comparar los resultados del grupo no aleatorizado de VELCADE en combinación con dexametasona (estudio Fase II etiqueta abierta MMY-2045), con resultados obtenidos en los brazos de tratamiento en monoterapia con VELCADE de diferentes ensayos aleatorizados de fase III (M34101-039 [APEX] y DOXIL MMY-3001) en la misma indicación.
El análisis de pares coincidentes es un método estadístico en el que los pacientes del grupo de tratamiento estudiado (por ejemplo, VELCADE en combinación con dexametasona) y los pacientes del grupo de comparación (por ejemplo, VELCADE) son comparables con respecto a los factores de confusión a través del emparejamiento individual de los sujetos del estudio. Este método minimiza los efectos de los factores de confusión observados al estimar los efectos del tratamiento utilizando datos no aleatorios.
Se identificaron 127 pares de pacientes emparejados. El análisis mostró una mejora en la tasa de respuesta general (ORR: CR + PR) (odds ratio 3,769; IC del 95%: 2,045-6,947; p
Hay información limitada disponible sobre el retratamiento con VELCADE en mieloma múltiple recidivante.
Se llevó a cabo el estudio de fase II MMY-2036 (RETRIEVE), de un solo brazo, abierto, para determinar la eficacia y seguridad del retratamiento con VELCADE. Ciento treinta pacientes (edad ≥ 18 años) con mieloma múltiple que previamente habían tenido al menos un tratamiento parcial La respuesta a un régimen que contenía VELCADE se retiró después de la progresión. Al menos 6 meses después de la terapia anterior, se inició VELCADE con la última dosis tolerada de 1,3 mg / m² (n = 93) o ≤ 1,0 mg / m² (n = 37) y se administró los días 1, 4, 8 y 11 cada 3 semanas durante un máximo de 8 ciclos, ya sea como monoterapia o en combinación con dexametasona según el estándar de terapia. Se administró dexametasona en combinación con VELCADE a 83 pacientes en el ciclo 1 y otros 11 pacientes recibieron dexametasona durante los siguientes ciclos de retratamiento de VELCADE.
El criterio de valoración principal fue la mejor respuesta confirmada al retratamiento según los criterios de EBMT. La mejor tasa de respuesta global (RC + PR) para el retratamiento en 130 pacientes fue del 38,5% (IC del 95%: 30,1; 47)., 4).
Eficacia clínica en pacientes con linfoma de células del manto (LCM) no tratados previamente
LYM-3002 es un estudio de fase III, aleatorizado y abierto que compara la eficacia y seguridad de la combinación de VELCADE, rituximab, ciclofosfamida, doxorrubicina y prednisona (VcR-CAP; n = 243) con los de rituximab. Ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP; n = 244) en pacientes adultos con MCL (estadio II, III o IV) no tratados previamente. Los pacientes del grupo de tratamiento con VcR-CAP recibieron VELCADE (1,3 mg / m²; los días 1, 4, 8, 11, período de descanso 12-21), rituximab 375 mg / m² IV el día 1; ciclofosfamida 750 mg / m² IV el día 1; doxorrubicina 50 mg / m² IV el día 1 y prednisona 100 mg / m² por vía oral los días 1 a 5 del ciclo de tratamiento de VELCADE de 21 días. Los pacientes con una primera respuesta documentada al ciclo 6 recibieron dos ciclos adicionales de tratamiento.
El criterio de valoración principal de eficacia fue la supervivencia libre de progresión según una evaluación del Comité de Revisión Independiente (IRC). Los criterios de valoración secundarios incluyeron el tiempo hasta la progresión (TTP), el tiempo hasta el próximo tratamiento antilinfoma (TNT), la duración del "intervalo sin tratamiento (TFI) ), tasa de respuesta global (ORR) y tasa de respuesta completa (CR / CRu), supervivencia global (SG) y duración de la respuesta.
Las características demográficas y basales de la enfermedad estuvieron generalmente bien equilibradas entre los dos grupos de tratamiento: la edad media de los pacientes fue de 66 años, el 74% eran hombres, el 66% eran caucásicos y el 32% asiáticos, el 69% de los pacientes. / o biopsia de médula ósea positiva para MCL, el 54% de los pacientes tenía una puntuación ≥ 3 en el Índice de pronóstico internacional (IPI) y el 76% tenía enfermedad en estadio IV. La duración del tratamiento (mediana = 17 semanas) y la duración del seguimiento (mediana = 40 meses) fueron comparables en ambos brazos de tratamiento. Los pacientes en ambos brazos de tratamiento recibieron una mediana de 6 ciclos con el 14% de los sujetos en el grupo VcR-CAP y el 17% de los pacientes en el grupo R-CHOP recibiendo los 2 ciclos adicionales. La mayoría de los pacientes de ambos grupos completaron el tratamiento, el 80% en el grupo VcR-CAP y el 82% en el grupo R-CHOP. Los resultados de eficacia se presentan en la Tabla 16:
Tabla 16: Resultados de eficacia del estudio LYM-3002
La mediana de SLP determinada por el investigador fue de 30,7 meses en el grupo VcR-CAP y de 16,1 meses en el grupo R-CHOP (Hazard Ratio [HR] = 0,51; p
La duración media de la respuesta completa fue de 42,1 meses en el grupo VcR-CAP en comparación con 18 meses en el grupo R-CHOP. La duración de la respuesta global fue 21,4 meses más larga en el grupo VcR-CAP (mediana de 36,5 meses frente a 15,1 meses en el grupo R-CHOP). Con una mediana de duración del seguimiento de 40 meses, la mediana de supervivencia global (SG) estuvo a favor de VcR-CAP (56,3 meses en el grupo R-CHOP y no se logró en el grupo VcR-CAP), (estimación de HR = 0,80; p = 0,173). Hubo una tendencia hacia una supervivencia global prolongada a favor del grupo VcR-CAP; la tasa de supervivencia estimada a 4 años fue del 53,9% en el grupo R-CHOP y del 64,4% en el grupo VcR-CAP.
Pacientes con amiloidosis de cadenas ligeras (AL) previamente tratada
Se realizó un estudio de fase I / II, abierto y no aleatorizado, para determinar la seguridad de VELCADE en pacientes con amiloidosis de cadenas ligeras (AL) previamente tratada. No se observaron nuevos problemas de seguridad durante el estudio y, en particular, VELCADE no provocó un empeoramiento del daño orgánico (corazón, riñón e hígado).
En un análisis exploratorio de eficacia, para las dos cohortes de dosis asociadas, se informó una tasa de respuesta del 67,3% (de la cual 28,6% de respuesta completa) en términos de respuesta hematológica (proteína M), en 49 pacientes evaluables tratados con las dosis máximas permitidas de 1,6 mg / m² una vez por semana y 1,3 mg / m² dos veces por semana Para los dos ciclos de dosis asociados, la supervivencia a 1 año fue del 88,1%.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los estudios con VELCADE en todos los subgrupos de la población pediátrica con mieloma múltiple y linfoma de células del manto (ver sección 4.2 para obtener información sobre el uso pediátrico).
05.2 "Propiedades farmacocinéticas -
Absorción
Después de la administración en bolo intravenoso de 1,0 mg / m² y 1,3 mg / m² a 11 pacientes con mieloma múltiple y valores de aclaramiento de creatinina superiores a 50 ml / min, las concentraciones plasmáticas máximas medias de bortezomib en las primeras dosis fueron 57 y 112 ng / ml. , respectivamente. En dosis posteriores, las concentraciones plasmáticas máximas medias observadas variaron de 67 a 106 ng / ml para la dosis de 1,0 mg / m² y entre 89 y 120 ng / ml para la dosis de 1,3 mg / m².
Después de repetidas inyecciones subcutáneas o en bolo intravenoso de una dosis de 1,3 mg / m² en pacientes con mieloma múltiple (n = 14 en el grupo intravenoso, n = 17 en el grupo subcutáneo), la exposición sistémica total al fármaco (AUClast) fue equivalente a la dosis subcutánea. y vías de administración intravenosa La Cmax tras la administración subcutánea (20,4 ng / ml) fue menor que la intravenosa (223 ng / ml) La media geométrica del AUC fue de 0,99 y con los intervalos de confianza del 90% fueron del 80,18% - 122,80%.
Distribución
En pacientes con mieloma múltiple, el volumen medio de distribución (Vd) de bortezomib osciló entre 1659 y 3294 l tras una dosis intravenosa única o repetida de 1,0 mg / m² o 1,3 mg / m². Esto sugiere que bortezomib se distribuye ampliamente en los tejidos periféricos. En un rango de concentración de bortezomib de 0,01 a 1,0 μg / ml, unión a proteínas plasmáticas humanas in vitro se situó en una media del 82,9%. La fracción de bortezomib unida a proteínas plasmáticas no dependió de la concentración.
Biotransformación
Educación in vitro en los microsomas hepáticos humanos y en las isoenzimas del citocromo P450 expresadas por el c-ADN humano indican que bortezomib experimenta principalmente un metabolismo oxidativo a través de las enzimas del citocromo P450, 3A4, 2C19 y 1A2. La principal vía metabólica consiste en la descoronación que conduce a dos metabolitos descoronados que posteriormente se hidroxilan a diferentes metabolitos. Los metabolitos sin corazón de bortezomib son inactivos como inhibidores del proteasoma 26S.
Eliminación
La vida media de eliminación media (t1 / 2) de bortezomib durante el tratamiento con dosis múltiples varía de 40 a 193 horas. Bortezomib se elimina más rápidamente después de la primera dosis que con las siguientes dosis. El aclaramiento total medio fue de 102 y 112 l / h después de la primera dosis de 1,0 mg / m² y 1,3 mg / m², respectivamente, y entre 15 y 32 l / hy entre 18 y 32 l / h para dosis posteriores de 1,0 mg / m² y 1,3 mg / m², respectivamente.
Poblaciones especiales
Insuficiencia hepática
El efecto de la insuficiencia hepática sobre la farmacocinética de bortezomib se estudió en un ensayo clínico de Fase I en 61 pacientes con tumores sólidos primarios con diversos grados de insuficiencia hepática y tratados con dosis de bortezomib entre 0,5 y 1,3 mg / m².
La insuficiencia hepática leve no alteró el AUC normalizado por dosis de bortezomib en comparación con el observado en pacientes con función hepática normal. Sin embargo, los valores medios de AUC normalizados por dosis aumentaron en aproximadamente un 60% en pacientes con insuficiencia hepática moderada o grave.Se recomienda una dosis inicial más baja en pacientes con insuficiencia hepática moderada o grave, y estos pacientes deben ser monitorizados cuidadosamente (ver sección 4.2, Tabla 6).
Insuficiencia renal
Se realizó un estudio farmacocinético en pacientes con diversos grados de insuficiencia renal, que se clasificaron según los valores de aclaramiento de creatinina (CrCL) en los siguientes grupos: Normal (CrCL ≥ 60 ml / min / 1,73 m², n = 12), Leve ( CrCL = 40-59 mL / min / 1.73 m², n = 10), Moderado (CrCL = 20-39 mL / min / 1.73 m², n = 9) y Severo (CrCL intravenoso en dosis que varían de 0.7 a 1.3 mg / m² dos veces por semana La exposición a VELCADE (dosis normalizada AUC y Cmax) fue similar en todos los grupos de pacientes (ver sección 4.2).
05.3 Datos preclínicos sobre seguridad -
En concentraciones
Bortezomib no mostró genotoxicidad en la prueba de mutagenicidad. in vitro (Prueba de Ames), ni en la prueba de micronúcleos en vivo llevado a cabo en ratones.
En estudios de toxicidad para el desarrollo realizados en ratas y conejos, se demostró mortalidad embriofetal a dosis tóxicas para la madre, pero ninguna toxicidad embriofetal por debajo de la dosis tóxica materna. No se han realizado estudios de fertilidad, sin embargo, se realizó una evaluación de los tejidos reproductivos en estudios de toxicidad general. En el estudio de seis meses en ratas, se encontraron efectos degenerativos tanto en los testículos como en los ovarios. Por tanto, es probable que bortezomib pueda tener un efecto potencial sobre la fertilidad masculina y femenina. No se han realizado estudios sobre el desarrollo perinatal y posnatal.
Los estudios de toxicidad general de ciclo múltiple llevados a cabo en ratas y monos revelaron que los principales órganos diana eran: el tracto gastrointestinal, lo que provocaba vómitos y / o diarrea; tejidos hematopoyéticos y linfáticos, que provocan citopenia en la sangre periférica, atrofia del tejido linfático e hipocelularidad hematopoyética de la médula ósea; neuropatía periférica (observada en monos, ratones y perros) que afecta a los axones de los nervios sensoriales; y ligeros cambios en el riñón. Después de la interrupción del tratamiento, todos estos órganos diana mostraron una recuperación parcial o completa.
Según estudios en animales, el paso de bortezomib a través de la barrera hematoencefálica parece limitado y se desconoce su relevancia en humanos.
Los estudios farmacológicos de seguridad cardiovascular realizados en monos y perros muestran que la administración intravenosa de dosis de mg / m2 de 2 a 3 veces la dosis clínicamente recomendada produce un aumento de la frecuencia cardíaca, disminución de la contractilidad cardíaca, hipotensión y muerte. En perros, la disminución de la contractilidad cardíaca y la hipotensión se controlaron mediante un tratamiento agudo con agentes inotrópicos positivos o vasopresores y se observó un ligero aumento en el intervalo QT corregido.
06.0 INFORMACIÓN FARMACÉUTICA -
06.1 Excipientes -
Manitol (E421)
Nitrógeno
06.2 Incompatibilidad "-
Este medicamento no debe mezclarse con otros productos excepto con los mencionados en la sección 6.6.
06.3 Período de validez "-
Vial sin abrir
3 años.
Solución reconstituida
La solución reconstituida debe usarse inmediatamente después de la preparación.
Si no se usa inmediatamente, es responsabilidad del usuario cumplir con las condiciones y tiempos de conservación del medicamento antes de su uso.
Sin embargo, se ha demostrado la estabilidad física y química de la solución reconstituida durante 8 horas a 25 ° C cuando se almacena en el vial original y / o en una jeringa. El tiempo total de conservación del medicamento reconstituido antes de la administración no debe exceder las 8 horas.
06.4 Precauciones especiales de conservación
Almacenar a una temperatura no superior a 30 ° C.
Mantenga el vial en el embalaje exterior para proteger el medicamento de la luz.
Para conocer las condiciones de almacenamiento después de la reconstitución del medicamento, ver sección 6.3.
06.5 Naturaleza del envase primario y contenido del envase.
Vial de vidrio tipo I de 10 ml, con tapón de bromobutilo gris y precinto de aluminio, con cápsula azul royal contiene 3,5 mg de bortezomib.
El vial está contenido en un blíster transparente que consta de una bandeja con tapa.
Cada paquete contiene 1 vial de un solo uso.
06.6 Instrucciones de uso y manipulación -
Precauciones generales
Bortezomib es un agente citotóxico. Por tanto, se debe tener especial cuidado al manipular y preparar VELCADE. Se recomienda usar guantes y otra ropa protectora para evitar el contacto con la piel.
La manipulación de VELCADE debe realizarse con estricto apego a técnicas asépticas debido a la ausencia de conservantes.
Ha habido casos de muerte tras la administración inadvertida de VELCADE por vía intratecal. VELCADE 1 mg polvo para solución inyectable está destinado únicamente para uso intravenoso, mientras que VELCADE 3,5 mg polvo para solución inyectable está destinado a uso intravenoso o subcutáneo VELCADE no debe administrarse por vía intratecal.
Instrucciones de reconstitución
VELCADE debe ser reconstituido por un profesional sanitario.
Inyección intravenosa
Cada vial de 10 ml que contiene VELCADE 3.5 polvo para solución inyectable debe reconstituirse con 3,5 ml de solución inyectable de cloruro sódico 9 mg / ml (0,9%). La disolución del polvo liofilizado tarda menos de 2 minutos. Después de la reconstitución, cada ml de solución contiene 1 mg de bortezomib. La solución reconstituida es transparente e incolora, con un pH final entre 4 y 7. La solución reconstituida debe inspeccionarse visualmente antes de la administración para comprobar si hay partículas o decoloración. En presencia de partículas o cambio de color. Coloree el reconstituido La solución no debe usarse y debe desecharse.
Inyección subcutánea
Cada vial de 10 ml que contiene VELCADE 3.5 polvo para solución inyectable debe reconstituirse con 1,4 ml de solución inyectable de cloruro sódico 9 mg / ml (0,9%). La disolución del polvo liofilizado tarda menos de 2 minutos. Después de la reconstitución, cada ml de solución contiene 2,5 mg de bortezomib. La solución reconstituida es transparente e incolora, con un pH final entre 4 y 7. La solución reconstituida debe inspeccionarse visualmente antes de la administración para comprobar si hay partículas o decoloración. En presencia de partículas o cambio de color. Coloree el reconstituido La solución no debe usarse y debe desecharse.
Disposición
VELCADE es para un solo uso.
Los medicamentos no utilizados y los desechos de este medicamento deben eliminarse de acuerdo con las regulaciones locales.
07.0 TITULAR DE LA "AUTORIZACIÓN DE COMERCIALIZACIÓN" -
JANSSEN-CILAG INTERNATIONAL N.V.
Turnhoutseweg, 30
B-2340 Beerse
Bélgica
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN -
EU / 1/04/274/001
036559019
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN -
Fecha de la primera autorización: 26 de abril de 2004
Fecha de la última renovación de la autorización: 26 de abril de 2014
10.0 FECHA DE REVISIÓN DEL TEXTO -
04/2015

















.jpg)



.jpg)