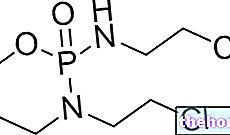
Ingredientes activos: Ácido zoledrónico
Zometa 4 mg polvo y disolvente para solución para perfusión
¿Por qué se usa Zometa? ¿Para qué sirve?
El principio activo de Zometa es el ácido zoledrónico, que pertenece a un grupo de sustancias llamadas bisfosfonatos. El ácido zoledrónico actúa uniéndose al hueso y ralentizando la velocidad del metabolismo. Se utiliza:
- Para prevenir complicaciones óseas, como fracturas, en pacientes adultos con metástasis óseas (diseminación del tumor desde el sitio del tumor primario hasta el hueso).
- Reducir la cantidad de calcio en sangre en pacientes adultos donde es demasiado alta debido a la presencia de un tumor. Los tumores pueden acelerar el metabolismo óseo normal de modo que aumente la liberación de calcio del hueso, condición que se conoce como hipercalcemia neoplásica (HIT).
Contraindicaciones Cuándo no se debe usar Zometa
Siga cuidadosamente todas las instrucciones que le haya dado su médico.
Antes de iniciar el tratamiento con Zometa, su médico le realizará análisis de sangre y comprobará su respuesta al tratamiento a intervalos regulares.
No se le debe administrar Zometa:
- si está amamantando.
- si es alérgico al ácido zoledrónico, a otro bisfosfonato (el grupo de sustancias al que pertenece Zometa) oa cualquiera de los demás componentes de este medicamento.
Precauciones de uso Lo que necesita saber antes de tomar Zometa
Hable con su médico antes de que le administren Zometa:
- si tiene o ha tenido alguna vez problemas de riñón.
- si tiene o ha tenido dolor, hinchazón o entumecimiento en la mandíbula o sensación de pesadez en la mandíbula o pérdida de un diente. Su médico puede recomendarle que se someta a un examen dental antes de comenzar el tratamiento con Zometa.
- Si está recibiendo tratamientos dentales o va a someterse a una cirugía dental, informe a su dentista que está siendo tratado con Zometa e informe a su médico sobre sus tratamientos dentales.
Durante el tratamiento con Zometa, debe mantener una buena higiene bucal (que incluye cepillarse los dientes con regularidad) y someterse a revisiones dentales de rutina. Informe a su médico y dentista de inmediato si experimenta algún problema con la boca o los dientes, como aflojamiento, dolor, hinchazón o llagas o secreciones que no cicatrizan, ya que estos pueden ser signos de una afección llamada osteonecrosis de la mandíbula.
Pacientes en quimioterapia y / o radioterapia, que toman esteroides, que se someten a cirugía dental, que no reciben atención dental de rutina, que tienen enfermedad de las encías, que son fumadores o que han sido tratados previamente con bisfosfonatos (utilizados para tratar o prevenir enfermedad ósea) tienen un mayor riesgo de desarrollar osteonecrosis de la mandíbula.
En pacientes tratados con Zometa, se han notificado niveles reducidos de calcio en sangre (hipocalcemia), que a veces pueden causar calambres musculares, piel seca, sensación de ardor. Se han notificado casos de latidos cardíacos irregulares (arritmia cardíaca), convulsiones, espasmos y contracciones musculares (tetania) secundarios a hipocalcemia grave. En algunas circunstancias, la hipocalcemia puede poner en peligro la vida. Si se encuentra en alguna de estas situaciones, informe a su médico de inmediato. Si existe una condición de hipocalcemia preexistente, debe tratarse antes de comenzar la primera dosis de Zometa. Se le administrará un suplemento adecuado de calcio y vitamina D.
Pacientes de 65 años y más
Zometa se puede administrar a personas mayores de 65 años. No hay evidencia de que sean necesarias precauciones adicionales.
Niños y adolescentes
No se recomienda el uso de Zometa en adolescentes y niños menores de 18 años.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Zometa?
Informe a su médico si está tomando, ha tomado recientemente o podría tomar cualquier otro medicamento. Es especialmente importante que informe a su médico si también está tomando:
- Aminoglucósidos (medicamentos utilizados para tratar infecciones graves), calcitonina (un tipo de medicamento utilizado para tratar la osteoporosis posmenopáusica y la hipercalcemia), diuréticos de asa (un tipo de medicamento utilizado para tratar la hipertensión arterial o el edema) u otros medicamentos que reducen los niveles de calcio, como La combinación de estas sustancias con bifosfonatos podría provocar una gran disminución de la concentración de calcio en sangre.
- Talidomida (un medicamento utilizado para tratar ciertos tipos de cánceres de la sangre que afectan los huesos) o cualquier otro medicamento que pueda ser perjudicial para los riñones.
- Aclasta (un medicamento que siempre contiene ácido zoledrónico y que se utiliza para tratar la osteoporosis y otras enfermedades óseas no cancerosas), o cualquier otro bisfosfonato, ya que se desconocen los efectos combinados de estos medicamentos cuando se toman junto con Zometa.
- Medicamentos antiangiogénicos (utilizados para tratar el cáncer), ya que la combinación de estos con Zometa se ha asociado con un mayor riesgo de osteonecrosis de la mandíbula.
Advertencias Es importante saber que:
Embarazo y lactancia
Si está embarazada, no se le debe administrar Zometa. Informe a su médico si está embarazada o sospecha de embarazo.
Si está amamantando, no se le debe administrar Zometa.
Pídale consejo a su médico antes de tomar cualquier medicamento durante el embarazo o si está amamantando.
Conducción y uso de máquinas
En muy raras ocasiones se han producido casos de somnolencia y somnolencia con el uso de Zometa, por lo que debe extremar las precauciones al conducir, utilizar maquinaria o realizar otras actividades que requieran una atención total.
Dosis, método y momento de administración Cómo usar Zometa: Posología
- Zometa solo debe ser administrado por profesionales sanitarios capacitados en el uso de bifosfonatos por vía intravenosa, es decir, a través de una vena.
- Su médico le recomendará que beba suficiente agua antes de cada tratamiento para ayudar a prevenir la deshidratación.
- Siga cuidadosamente todas las demás instrucciones dadas por su médico, farmacéutico o enfermero.
Cuanto se administra
- La dosis única habitual es de 4 mg.
- Si tiene problemas de riñón, su médico le dará una dosis reducida según la gravedad del problema de riñón.
¿Con qué frecuencia se administra Zometa?
- Si está recibiendo tratamiento para la prevención de complicaciones óseas causadas por metástasis óseas, se le administrará una infusión de Zometa cada tres o cuatro semanas.
- Si está siendo tratado para reducir la cantidad de calcio en sangre, normalmente solo se le administrará una perfusión de Zometa.
Cómo se administra Zometa
- Zometa se administra en una vena como una perfusión que dura al menos 15 minutos y debe administrarse como una única solución intravenosa en una línea de perfusión separada.
A los pacientes cuyos niveles de calcio en sangre no sean demasiado altos también se les prescribirán suplementos de calcio y vitamina D, que se tomarán todos los días.
Sobredosis Qué hacer si ha tomado demasiado Zometa
Si le han administrado dosis superiores a las recomendadas, su médico debe controlarlo cuidadosamente. Esto se debe a que puede desarrollar anomalías en los electrolitos séricos (por ejemplo, niveles anormales de calcio, fósforo y magnesio) y / o cambios en la función renal, incluyendo insuficiencia renal grave. Si sus niveles de calcio bajan demasiado, es posible que deba administrarle suplementos de calcio por infusión.
Efectos secundarios ¿Cuáles son los efectos secundarios de Zometa?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los más comunes suelen ser leves y probablemente desaparecerán en poco tiempo.
Informe a su médico si presenta alguno de los siguientes efectos secundarios graves:
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes):
- Insuficiencia renal grave (esto lo comprobará su médico mediante algunos análisis de sangre específicos).
- Niveles bajos de calcio en sangre.
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes):
- Dolor en la boca, dientes y / o mandíbula, hinchazón o llagas que no cicatrizan dentro de la boca o mandíbula, secreción, entumecimiento o sensación de pesadez en la mandíbula, o aflojamiento de un diente. Ser signos de una lesión en la mandíbula. (osteonecrosis). Si experimenta estos síntomas durante o después de interrumpir el tratamiento con Zometa, informe a su médico y dentista inmediatamente.
- Se ha observado un ritmo cardíaco irregular (fibrilación auricular) en pacientes que están siendo tratadas con ácido zoledrónico para la osteoporosis posmenopáusica.Actualmente se desconoce si el ácido zoledrónico causa este ritmo cardíaco irregular, pero debe informar a su médico si presenta estos síntomas después de su tratamiento. administrado ácido zoledrónico.
- Reacciones alérgicas graves: dificultad para respirar, hinchazón especialmente de la cara y la garganta.
Raras (pueden afectar hasta 1 de cada 1.000 pacientes):
- Como consecuencia de niveles bajos de calcio: latidos cardíacos irregulares (arritmia cardíaca secundaria a hipocalcemia).
Muy raros (pueden afectar hasta 1 de cada 10.000 pacientes):
- Como consecuencia de niveles bajos de calcio: convulsiones, entumecimiento y tetania (secundaria a hipocalcemia).
Informe a su médico lo antes posible si presenta alguno de los siguientes efectos secundarios:
Muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes):
- Niveles bajos de fosfatos en sangre.
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes):
- Dolor de cabeza y un síndrome similar a la gripe con fiebre, fatiga, debilidad, somnolencia, escalofríos y dolores en los huesos, articulaciones y / o músculos. En la mayoría de los casos, no se requiere un tratamiento específico y los síntomas desaparecen al poco tiempo (un par de horas o días).
- Reacciones gastrointestinales, como náuseas y vómitos, así como pérdida de apetito.
- Conjuntivitis.
- Niveles bajos de glóbulos rojos (anemia).
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes):
- Reacciones hipersensibles.
- Presión arterial baja.
- Dolor de pecho.
- Reacciones cutáneas (enrojecimiento e hinchazón) en el lugar de la perfusión, erupción cutánea, picor.
- Presión arterial alta, dificultad para respirar, mareos, ansiedad, alteraciones del sueño, alteraciones del gusto, temblores, hormigueo o entumecimiento de manos o pies, diarrea, estreñimiento, dolor abdominal, sequedad de boca.
- Niveles bajos de glóbulos blancos y plaquetas en la sangre.
- Niveles bajos de magnesio y potasio en sangre. El médico los controlará y tomará las medidas necesarias.
- Aumento de peso.
- Aumento de la sudoración.
- Somnolencia.
- Visión borrosa, daño ocular, sensibilidad a la luz.
- Escalofríos repentinos con desmayos, debilidad o colapso.
- Dificultad para respirar con sibilancias o tos.
- Urticaria.
Raras (pueden afectar hasta 1 de cada 1.000 pacientes):
- Latido lento.
- Confusión.
- En raras ocasiones, puede producirse una fractura inusual del fémur, especialmente en pacientes en tratamiento a largo plazo para la osteoporosis. Comuníquese con su médico si experimenta dolor, debilidad o malestar en el muslo, la cadera o la ingle, ya que esto puede ser una indicación temprana. De una posible fractura del fémur.
- Enfermedad pulmonar intersticial (inflamación del tejido pulmonar).
- Síntomas similares a los de la gripe que incluyen artritis e inflamación de las articulaciones.
- Enrojecimiento doloroso y / o hinchazón de los ojos.
Muy raros (pueden afectar hasta 1 de cada 10.000 pacientes):
- Desmayos debido a la presión arterial baja.
- Dolor severo en los huesos, articulaciones y / o músculos, ocasionalmente incapacitante.
Notificación de efectos secundarios
Si experimenta cualquier efecto adverso, consulte a su médico, farmacéutico o enfermero. Esto incluye cualquier posible efecto adverso que no se mencione en este prospecto. También puede notificar los efectos secundarios directamente a través del sistema nacional de notificación incluido en el Apéndice V.Efectos secundarios que puede ayudar proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
Su médico, farmacéutico o enfermero saben cómo conservar correctamente Zometa (ver sección 6).
Qué contiene Zometa
- El principio activo de Zometa es ácido zoledrónico Un vial contiene 4 mg de ácido zoledrónico, correspondientes a 4,264 mg de ácido zoledrónico monohidrato.
- Los demás componentes son: manitol, citrato de sodio.
Aspecto de Zometa y contenido del envase
Zometa se presenta en forma de polvo en un vial. Un vial contiene 4 mg de ácido zoledrónico.
Cada envase contiene el vial de polvo con una ampolla de 5 ml de agua para preparaciones inyectables que se utilizará para disolver el polvo.
Zometa se presenta en envases individuales que contienen 1 o 40 viales y 1 o 4 ampollas respectivamente y en envases múltiples que contienen 10 (10x 1 + 1) viales y ampollas.
Es posible que no se comercialicen todos los tamaños de envases.
INFORMACIÓN PARA EL PERSONAL DE SALUD
Cómo preparar y administrar Zometa
- Para preparar una solución para perfusión que contenga 4 mg de ácido zoledrónico, añadir, en condiciones estériles, 5 ml de agua para preparaciones inyectables, utilizando el vial especial incluido en el envase del producto, al vial que contiene el polvo de Zometa. Agitar el vial suavemente. disolver el polvo.
- Diluya más la solución reconstituida de Zometa (5 ml) con 100 ml de solución para perfusión sin calcio u otro catión divalente. Si se requiere una dosis reducida de Zometa, primero extraiga el volumen apropiado de solución reconstituida (4 mg / 5 ml) como se indica a continuación y luego diluya en 100 ml de solución para perfusión. Para evitar posibles incompatibilidades, la solución para perfusión utilizada para la dilución debe ser una solución salina al 0,9% p / v o una solución de glucosa al 5% p / v.
La solución reconstituida de Zometa no debe mezclarse con soluciones que contengan calcio u otros cationes divalentes como la solución de lactato de Ringer.
Instrucciones para la preparación de Zometa en dosis reducidas:
Extraiga el volumen apropiado de la solución reconstituida (4 mg / 5 ml), como se indica a continuación:
- 4,4 ml para la dosis de 3,5 mg
- 4,1 ml para la dosis de 3,3 mg
- 3,8 ml para la dosis de 3,0 mg
- Para un solo uso.
Cualquier resto de solución no utilizada debe desecharse. Solo se debe utilizar la solución transparente, libre de partículas visibles e incolora. Deben seguirse técnicas asépticas durante la preparación de la perfusión.
- Desde un punto de vista microbiológico, la solución para perfusión reconstituida y diluida debe utilizarse inmediatamente después de la primera apertura. Si no se usa inmediatamente, los tiempos de almacenamiento en uso y las condiciones antes de su uso son responsabilidad del usuario y normalmente no deberían exceder las 24 horas a 2 ° C - 8 ° C.La solución refrigerada debe devolverse. A temperatura ambiente antes de su uso. administración.
- La solución que contiene ácido zoledrónico debe administrarse como una única perfusión que dure 15 minutos en una línea de perfusión separada. Se debe evaluar el estado de hidratación de los pacientes antes y después de la administración de Zometa para asegurarse de que estén adecuadamente hidratados.
- Los estudios realizados en diferentes líneas de perfusión compuestas por cloruro de polivinilo, polietileno y polipropileno no han mostrado incompatibilidad con Zometa.
- Como no hay datos sobre la compatibilidad de Zometa con otras sustancias administradas por vía intravenosa, Zometa no debe mezclarse con otros medicamentos y / o sustancias y siempre debe administrarse a través de una línea de perfusión separada.
Conservación de Zometa
- Mantenga Zometa fuera de la vista y del alcance de los niños.
- No use Zometa después de la fecha de caducidad que se indica en el paquete.
- El vial cerrado no requiere condiciones especiales de conservación.
- La solución para perfusión de Zometa diluida debe utilizarse inmediatamente para evitar la contaminación microbiológica.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO
ZOMETA 4 MG POLVO Y DISOLVENTE PARA SOLUCIÓN PARA INFUSIÓN
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene 4 mg de ácido zoledrónico, correspondientes a 4,264 mg de ácido zoledrónico monohidrato.
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA
Polvo y disolvente para solución para perfusión.
Polvo blanco a blanquecino y transparente, disolvente incoloro.
04.0 INFORMACIÓN CLÍNICA
04.1 Indicaciones terapéuticas
• Prevención de eventos relacionados con el esqueleto (fracturas patológicas, aplastamiento vertebral, radioterapia o cirugía ósea, hipercalcemia neoplásica) en pacientes adultos con tumores malignos avanzados que afectan al hueso.
• Tratamiento de pacientes adultos con hipercalcemia neoplásica (HIT).
04.2 Posología y forma de administración
Zometa solo debe ser prescrito y administrado a pacientes por profesionales sanitarios con experiencia en la administración de bisfosfonatos intravenosos. Los pacientes tratados con Zometa deben recibir el prospecto y la tarjeta recordatorio para el paciente.
Dosis
Prevención de eventos relacionados con el esqueleto en pacientes con tumores malignos avanzados que afectan al hueso
Adultos y ancianos
La dosis recomendada para la prevención de eventos relacionados con el esqueleto en pacientes con neoplasias avanzadas que afectan al hueso es de 4 mg de ácido zoledrónico cada 3 a 4 semanas.
Los pacientes también deben recibir un suplemento de 500 mg / día de calcio oral y 400 UI / día de vitamina D.
La decisión de tratar a pacientes con metástasis óseas para la prevención de eventos esqueléticos relacionados debe tener en cuenta que el efecto del tratamiento se manifiesta en 2-3 meses.
Tratamiento de TIH
Adultos y ancianos
La dosis recomendada en la hipercalcemia (calcio corregido con albúmina ≥ 12,0 mg / dl o 3,0 mmol / l) es una dosis única de 4 mg de ácido zoledrónico.
Pacientes con insuficiencia renal
TIH:
En pacientes con HIT que también tienen insuficiencia renal grave, el tratamiento con Zometa solo debe considerarse después de evaluar los riesgos y beneficios del tratamiento. En los ensayos clínicos, se excluyó a los pacientes con creatinina sérica> 400 μmol / L o> 4,5 mg / dL. No se requiere ajuste de dosis en pacientes con HIT con valores de creatinina sérica.
Prevención de eventos relacionados con el esqueleto en pacientes con tumores malignos avanzados:
Se debe determinar la creatinina sérica y el aclaramiento de creatinina (CLcr) antes de iniciar el tratamiento con Zometa en pacientes con mieloma múltiple o metástasis óseas de tumores sólidos. El CLcr se calcula a partir de la creatinina sérica utilizando la fórmula de Cockcroft-Gault. No se recomienda Zometa en pacientes que presenten insuficiencia renal grave antes del inicio del tratamiento, definido para esta población como CLcr 265 μmol / lo> 3,0 mg / dl.
En pacientes con metástasis óseas con insuficiencia renal leve a moderada, definida para esta población como CLcr 30-60 ml / min, se recomienda la siguiente dosis de Zometa (ver también sección 4.4):
* Las dosis se calcularon asumiendo un AUC objetivo de 0,66 (mg • h / L) (CLcr = 75 ml / min). Con la administración de la dosis reducida en pacientes con insuficiencia renal, se espera alcanzar un valor de AUC igual al observado en pacientes con un aclaramiento de creatinina de 75 ml / min.
Después del inicio del tratamiento, se debe determinar la creatinina sérica antes de cada administración de Zometa y, en caso de empeoramiento de la función renal, se debe interrumpir el tratamiento.En los estudios clínicos, el empeoramiento de la función renal se definió de la siguiente manera:
- Para pacientes con valores basales de creatinina sérica normales (
• Para pacientes con valores basales de creatinina sérica anormales (> 1,4 mg / dL o> 124 μmol / L), un aumento de 1,0 mg / dL o 88 μmol / L.
En los estudios clínicos, el tratamiento con Zometa se reanudó solo cuando la creatinina volvió a un 10% del valor inicial (ver sección 4.4). El tratamiento con Zometa debe reanudarse con la misma concentración que se utilizó antes de interrumpir el tratamiento.
Población pediátrica
No se ha establecido la seguridad y eficacia del ácido zoledrónico en niños de 1 a 17 años. Los datos actualmente disponibles se describen en la sección 5.1, pero no se puede hacer una recomendación posológica.
Método de administración
Vía intravenosa.
Zometa 4 mg polvo y disolvente para solución para perfusión, reconstituidos y posteriormente diluidos a 100 ml (ver sección 6.6) deben administrarse como una única perfusión intravenosa durante no menos de 15 minutos.
En pacientes con insuficiencia renal leve o moderada, se recomienda una reducción de la dosis de Zometa (ver sección "Posología" anterior y sección 4.4).
Instrucciones para preparar Zometa en dosis reducidas
Extraiga, según sea necesario, un volumen adecuado de la solución reconstituida (4 mg / 5 ml):
- 4,4 ml para la dosis de 3,5 mg
- 4,1 ml para la dosis de 3,3 mg
• 3,8 ml para la dosis de 3,0 mg
Para obtener instrucciones sobre la reconstitución y dilución del medicamento antes de la administración, ver sección 6.6. La cantidad extraída de la solución reconstituida debe diluirse en 100 ml de solución salina estéril al 0,9% p / v o solución de glucosa al 5% p / v. La dosis debe administrarse como una única perfusión intravenosa que dure no menos de 15 minutos.
La solución reconstituida de Zometa no debe mezclarse con soluciones para perfusión que contengan calcio u otros cationes divalentes como la solución de lactato de Ringer, y debe administrarse como una única solución intravenosa en una línea de perfusión separada.
Los pacientes deben mantenerse bien hidratados antes y después de la administración de Zometa.
04.3 Contraindicaciones
• Hipersensibilidad al principio activo, a otros bifosfonatos oa alguno de los excipientes incluidos en la sección 6.1.
• Lactancia materna (ver sección 4.6)
04.4 Advertencias especiales y precauciones de uso apropiadas
General
Antes de la administración de Zometa, los pacientes deben ser evaluados cuidadosamente para asegurarse de que estén adecuadamente hidratados.
Debe evitarse la hidratación excesiva en pacientes con riesgo de insuficiencia cardíaca.
Durante el tratamiento con Zometa, deben vigilarse estrechamente los parámetros metabólicos normales relacionados con la hipercalcemia, como los niveles séricos de calcio, fosfato y magnesio. Si se produce hipocalcemia, hipofosfatemia o hipomagnesemia, puede ser necesario un tratamiento complementario a corto plazo. Los pacientes con hipercalcemia no tratada generalmente tienen cierto grado de insuficiencia renal, por lo que se debe considerar una monitorización cuidadosa de la función renal.
Zometa contiene el mismo principio activo que Aclasta (ácido zoledrónico). Los pacientes tratados con Zometa no deben ser tratados simultáneamente con Aclasta o cualquier otro bisfosfonato, ya que se desconoce el efecto combinado de estos agentes.
Insuficiencia renal
Los pacientes con HIT y que presenten signos de empeoramiento de la función renal deben ser evaluados adecuadamente, considerando si los beneficios potenciales del tratamiento con Zometa superan los riesgos.
La decisión de tratar a pacientes con metástasis óseas para la prevención de eventos relacionados con el esqueleto debe tener en cuenta el hecho de que el efecto del tratamiento comienza a manifestarse después de 2-3 meses.
El tratamiento con Zometa se ha asociado con informes de trastornos de la función renal. Los factores que pueden aumentar el riesgo de empeoramiento de la función renal incluyen deshidratación, insuficiencia renal preexistente, ciclos múltiples de Zometa y otros bifosfonatos, así como el uso de otros fármacos nefrotóxicos. Aunque el riesgo se reduce con la administración de 4 mg de ácido zoledrónico durante 15 minutos, sin embargo, puede ocurrir un empeoramiento de la función renal. Se ha informado de empeoramiento de la función renal, progresión a insuficiencia renal y diálisis en pacientes después de la primera dosis o después de una dosis única de 4 mg de ácido zoledrónico. También se puede observar un aumento de la creatinina sérica en algunos pacientes cuando se administra Zometa a largo plazo y en la dosis recomendada para la prevención de eventos relacionados con el esqueleto, aunque estos casos son menos frecuentes.
Se deben evaluar los niveles de creatinina sérica del paciente antes de la administración de cada dosis de Zometa. Se recomienda que el tratamiento con ácido zoledronio se inicie a dosis reducidas en pacientes con metástasis óseas con insuficiencia renal leve a moderada. En pacientes que muestren signos de insuficiencia renal durante el tratamiento, se debe interrumpir el tratamiento con Zometa. Zometa solo debe reiniciarse cuando el valor de creatinina sérica regrese dentro del 10% del valor inicial. El tratamiento con Zometa debe reanudarse con la misma concentración que se utilizó antes de interrumpir el tratamiento.
En vista del posible impacto del ácido zoledrónico en la función renal, la falta de datos clínicos de seguridad en pacientes con insuficiencia renal grave (definida en los ensayos clínicos como creatinina sérica ≥ 400 μmol / lo ≥ 4,5 mg / dl para pacientes con HIT y ≥ 265 μmol / L o ≥ 3,0 mg / dL para pacientes con cáncer y metástasis óseas) al inicio y datos farmacocinéticos limitados en pacientes con insuficiencia renal grave al inicio (aclaramiento de creatinina
Insuficiencia hepática
Dado que los datos clínicos disponibles en pacientes con insuficiencia hepática grave son limitados, no es posible hacer recomendaciones específicas en esta población de pacientes.
Osteonecrosis de la mandíbula / maxilar
Se ha notificado osteonecrosis de la mandíbula como un evento poco común en los ensayos clínicos y en el período poscomercialización en pacientes que recibieron Zometa.
El inicio del tratamiento o un nuevo ciclo de tratamiento debe posponerse en pacientes con lesiones abiertas de tejidos blandos de la cavidad bucal que no hayan cicatrizado, excepto en situaciones de emergencia médica.Antes de iniciar el tratamiento con bisfosfonatos en pacientes con factores de riesgo concomitantes, se recomienda un examen dental con los procedimientos dentales preventivos adecuados y una evaluación individual del beneficio-riesgo.
Se deben considerar los siguientes factores de riesgo al evaluar el riesgo individual de desarrollar osteonecrosis de la mandíbula:
• potencia del bisfosfonato (mayor riesgo para productos con mayor potencia), vía de administración (mayor riesgo para la administración parenteral) y dosis acumulativa de bisfosfonato.
• cáncer, comorbilidades (p. Ej. Anemia, coagulopatías, infección), tabaquismo.
• terapias concomitantes: quimioterapia, inhibidores de la angiogénesis (ver sección 4.5), radioterapia en el cuello y la cabeza, corticosteroides.
• antecedentes de enfermedad dental, mala higiene bucal, enfermedad periodontal, procedimientos dentales invasivos (por ejemplo, extracciones dentales) y dentaduras postizas mal ajustadas.
Se debe alentar a todos los pacientes a que mantengan una buena higiene bucal, se sometan a controles dentales de rutina y notifiquen inmediatamente cualquier síntoma bucal como movilidad de los dientes, dolor, hinchazón o llagas que no cicatrizan o secreciones durante el tratamiento con Zometa. Durante el tratamiento, los procedimientos dentales invasivos solo deben realizarse después de una cuidadosa consideración y evitarse en las proximidades de la administración de ácido zoledrónico. En pacientes que han desarrollado osteonecrosis de la mandíbula durante el tratamiento con bisfosfonatos, la cirugía dental puede exacerbar la afección. requiriendo cirugía dental, no hay datos disponibles que sugieran que la interrupción del tratamiento con bisfosfonatos reduzca el riesgo de osteonecrosis de la mandíbula.
El programa de tratamiento para los pacientes que desarrollan osteonecrosis de la mandíbula debe establecerse en estrecha colaboración entre el médico tratante y un dentista o cirujano oral competente en la osteonecrosis de la mandíbula. Se debe considerar la interrupción temporal del tratamiento con ácido zoledrónico hasta que la afección se resuelva y los factores de riesgo concomitantes se mitiguen cuando sea posible.
Dolor musculoesquelético
Durante la experiencia postcomercialización, se han notificado casos de dolor óseo, articular y / o muscular grave y ocasionalmente incapacitante en pacientes tratados con Zometa. Sin embargo, estos informes fueron infrecuentes. Los síntomas variaron de un día a varios meses. La mayoría de los pacientes experimentaron "alivio de los síntomas después de suspender" el tratamiento. Un subgrupo experimentó una recaída de los síntomas al recibir tratamiento adicional con Zometa u otro bifosfonato.
Fracturas atípicas del fémur.
Se han notificado fracturas subtrocantéreas y diafisarias atípicas del fémur, principalmente en pacientes que reciben tratamiento prolongado con bisfosfonatos para la osteoporosis. Estas fracturas cortas transversales u oblicuas pueden ocurrir en cualquier parte del fémur, desde justo debajo del trocánter menor hasta por encima de la línea supracondílea. Estas fracturas ocurren espontáneamente o después de un trauma mínimo y algunos pacientes experimentan dolor en el muslo o la ingle, a menudo asociado con evidencia de imágenes de fracturas por estrés, semanas o meses antes de que ocurra una fractura de cadera. Las fracturas suelen ser bilaterales; por lo tanto, en pacientes tratados con bisfosfonatos que han sufrido una fractura de la diáfisis femoral, debe examinarse el fémur contralateral. También se ha informado de una curación limitada de estas fracturas. En pacientes con sospecha de fractura atípica de fémur, se debe considerar la interrupción del tratamiento con bifosfonatos en espera de una evaluación del paciente basada en el beneficio-riesgo individual.
Durante el tratamiento con bifosfonatos, se debe advertir a los pacientes que notifiquen cualquier dolor en el muslo, la cadera o la ingle y se debe evaluar a cualquier paciente que presente estos síntomas en busca de una fractura incompleta del fémur.
Hipocalcemia
Se han notificado casos de hipocalcemia en pacientes tratados con Zometa. Se han notificado arritmias cardíacas y reacciones adversas neurológicas (incluidas convulsiones, hipoestesia y tetania) secundarias a casos de hipocalcemia grave. Se han notificado casos de hipocalcemia grave que requirieron hospitalización. En algunas circunstancias, la hipocalcemia puede poner en peligro la vida (ver sección 4.8). Se recomienda especial precaución cuando se administra Zometa con medicamentos que se sabe que causan hipocalcemia, ya que pueden tener un efecto sinérgico dando lugar a una hipocalcemia grave (ver sección 4.5) Calcio sérico. debe medirse y tratarse la hipocalcemia antes de iniciar el tratamiento con Zometa. Los pacientes deben recibir "una suplementación adecuada de calcio y vitamina D".
04.5 Interacciones con otros medicamentos y otras formas de interacción
En los estudios clínicos, Zometa se administró de forma concomitante con los fármacos anticancerosos, diuréticos, antibióticos y analgésicos de uso común, sin que se hayan observado interacciones clínicamente significativas. In vitro Se ha demostrado que el ácido zoledrónico no se une a las proteínas plasmáticas y no inhibe las enzimas del citocromo P450 (ver sección 5.2), pero no se han realizado estudios de interacción clínica específicos con otros medicamentos.
Se recomienda especial precaución si se administran bisfosfonatos con aminoglucósidos, calcitonina o diuréticos del asa, ya que estos medicamentos pueden tener un efecto aditivo que resulte en una disminución del calcio durante períodos más prolongados de lo necesario (ver sección 4.4).
Se recomienda precaución cuando se administra Zometa con otros medicamentos potencialmente nefrotóxicos. También preste atención a la posible aparición de hipomagnesemia durante el tratamiento.
En pacientes con mieloma múltiple, el riesgo de disfunción renal puede aumentar cuando se usa Zometa en combinación con talidomida.
Se debe tener precaución cuando se administre Zometa con medicamentos antiangiogénicos, ya que se ha observado una mayor incidencia de casos de ONM en pacientes tratados concomitantemente con estos medicamentos.
04.6 Embarazo y lactancia
El embarazo
No existen datos suficientes sobre el uso de ácido zoledrónico en mujeres embarazadas. Los estudios de reproducción con ácido zoledrónico en animales han mostrado toxicidad reproductiva (ver sección 5.3). Se desconoce el riesgo potencial para los seres humanos. Zometa no debe usarse durante el embarazo Mujeres de Se debe advertir sobre el potencial fértil para evitar quedar embarazada.
El embarazo
Se desconoce si el ácido zoledrónico se excreta en la leche materna. Zometa está contraindicado en mujeres en período de lactancia (ver sección 4.3).
Fertilidad
El ácido zoledrónico fue estudiado en ratas por posibles efectos adversos sobre la fertilidad de los padres y la generación F1. Mostró efectos farmacológicos muy evidentes considerados relacionados con la inhibición del compuesto sobre el metabolismo del calcio esquelético, resultando en hipocalcemia en el periparto, un efecto de clase de bisfosfonatos, distocia y cierre temprano del estudio. Por esta razón, estos resultados impidieron la determinación definitiva de los efectos del ácido zoledrónico en la fertilidad humana.
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Las reacciones adversas, como mareos y somnolencia, pueden afectar a la capacidad para conducir o utilizar máquinas, por lo que se debe tener precaución al conducir y utilizar máquinas durante el tratamiento con Zometa.
04.8 Efectos indeseables
Resumen del perfil de seguridad
En los tres días posteriores a la administración de Zometa, se informó comúnmente una reacción de fase aguda, con síntomas que incluían dolor de huesos, fiebre, fatiga, artralgia, mialgia, rigidez y artritis que producían hinchazón de las articulaciones; estos síntomas generalmente se resolvieron en unos pocos días (ver descripción de eventos adversos seleccionados).
Los siguientes han sido identificados como riesgos importantes con el uso de Zometa en las indicaciones aprobadas:
insuficiencia renal, osteonecrosis de la mandíbula, reacción de fase aguda, hipocalcemia, fibrilación auricular, anafilaxia y enfermedad pulmonar intersticial. Las frecuencias para cada uno de estos riesgos identificados se muestran en la Tabla 1.
Tabla de reacciones adversas
Las siguientes reacciones adversas, enumeradas en la Tabla 1, se derivaron de estudios clínicos e informes posteriores a la comercialización tras la administración crónica de 4 mg de ácido zoledrónico:
tabla 1
Las reacciones adversas se clasifican en orden descendente de frecuencia utilizando la siguiente convención: Muy frecuentes (≥1 / 10), frecuentes (≥1 / 100,
Descripción de reacciones adversas seleccionadas
Insuficiencia renal
Zometa se ha asociado con informes de disfunción renal. En un análisis de los datos de seguridad agrupados de los estudios fundamentales de Zometa en la prevención de acontecimientos relacionados con el esqueleto en pacientes con neoplasias malignas avanzadas que afectan al hueso, se determinó la frecuencia de acontecimientos adversos de insuficiencia renal que se sospecha están relacionados con el uso de Zometa (reacciones adversas). los siguientes: mieloma múltiple (3,2%), cáncer de próstata (3,1%), cáncer de mama (4,3%), cáncer de pulmón y otros tumores sólidos (3, 2%). Los factores que pueden aumentar la posibilidad de empeoramiento de la función renal incluyen deshidratación, insuficiencia renal preexistente, ciclos múltiples de Zometa u otros bifosfonatos, así como el uso concomitante de fármacos nefrotóxicos o un tiempo de perfusión más corto de lo que generalmente se recomienda. Insuficiencia renal, progresión a Se han notificado casos de insuficiencia renal y diálisis en pacientes tras la dosis inicial o una dosis única de 4 mg de ácido zoledrónico (ver sección 4.4).
Osteonecrosis de la mandíbula / maxilar
Se han notificado casos de osteonecrosis de la mandíbula, principalmente en pacientes con cáncer tratados con medicamentos que inhiben la resorción ósea, como Zometa (ver sección 4.4). Muchos de estos pacientes también estaban recibiendo quimioterapia y corticosteroides y tenían evidencia de infección localizada, incluida la osteomielitis. La mayoría de los informes se refieren a pacientes con cáncer que se someten a extracciones dentales u otras cirugías dentales.
Fibrilación auricular
En un estudio controlado, aleatorizado, doble ciego de 3 años que evaluó la eficacia y seguridad de 5 mg de ácido zoledrónico una vez al año frente a placebo en el tratamiento de la osteoporosis posmenopáusica (OPM), la incidencia global de fibrilación auricular fue del 2,5% (96 de 3.862) y 1.9% (75 de 3.852) en pacientes que recibieron ácido zoledrónico 5 mg y placebo, respectivamente. La tasa de eventos adversos graves de fibrilación auricular fue respectivamente del 1.3% (51 de 3.862) y 0.6% (22 de 3.852). El desequilibrio observado en este estudio no se observó en otros estudios con ácido zoledrónico, incluidos aquellos con Zometa (ácido zoledrónico) 4 mg cada 3-4 semanas en pacientes con cáncer. Se desconoce el mecanismo subyacente al aumento de la incidencia de fibrilación auricular en este único estudio. .
Reacción de fase aguda
Esta reacción adversa al medicamento incluye una variedad de síntomas que incluyen fiebre, mialgia, dolor de cabeza, dolor en las extremidades, náuseas, vómitos, diarrea, artralgia y artritis que provocan inflamación de las articulaciones. El tiempo de aparición es ≤ 3 días después de la infusión de Zometa y la reacción también se conoce como "síntomas similares a la gripe" o síntomas "posteriores a la dosis".
Fracturas atípicas de fémur
Se han notificado las siguientes reacciones durante la experiencia postcomercialización (frecuencia rara):
Fracturas subtrocantéreas y diafisarias atípicas del fémur (reacción adversa de clase de bisfosfonatos).
Reacciones adversas (RAM) relacionadas con la hipocalcemia
La hipocalcemia es un riesgo importante identificado con Zometa en las indicaciones aprobadas. Según la revisión de casos tanto de ensayos clínicos como del uso poscomercialización, existe evidencia suficiente para apoyar una asociación entre el tratamiento con Zometa, los eventos notificados de hipocalcemia y el desarrollo secundario de problemas cardíacos. arritmia.También existe evidencia de una asociación entre hipocalcemia y eventos neurológicos secundarios notificados en estos casos, incluyendo convulsiones, hipoestesia y tetania (ver sección 4.4).
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas que se produzcan después de la autorización del medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento. Se ruega a los profesionales sanitarios que notifiquen cualquier sospecha de reacciones adversas a través del sistema nacional de notificación.
04.9 Sobredosis
La experiencia clínica con sobredosis aguda de Zometa es limitada. Se ha notificado la administración errónea de dosis de hasta 48 mg de ácido zoledrónico. Se debe monitorizar a los pacientes que han sido tratados con dosis superiores a las recomendadas (ver sección 4.2). Se ha observado insuficiencia (incluyendo insuficiencia renal) y alteraciones de los electrolitos séricos (incluyendo calcio, fósforo y magnesio) En caso de hipocalcemia, se deben administrar perfusiones de gluconato de calcio según esté clínicamente indicado.
05.0 PROPIEDADES FARMACOLÓGICAS
05.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Medicamentos para el tratamiento de enfermedades óseas, bisfosfonatos, código ATC: M05BA08.
El ácido zoledrónico pertenece a la clase de los bisfosfonatos y actúa principalmente a nivel óseo. Es un inhibidor de la absorción ósea osteoclástica.
La acción selectiva de los bifosfonatos sobre el tejido óseo se debe a su alta afinidad por el hueso mineralizado, pero aún no se conoce el mecanismo molecular exacto que determina la inhibición de la actividad osteoclástica. Estudios a largo plazo en animales han demostrado que el ácido zoledrónico inhibe la resorción ósea sin efectos adversos que afectan la formación ósea, la mineralización o las propiedades mecánicas.
Además de ser un potente inhibidor de la resorción ósea, el ácido zoledrónico también posee varias propiedades anticancerígenas que podrían contribuir a su eficacia general en el tratamiento de metástasis óseas. Las siguientes propiedades se han demostrado en estudios preclínicos:
• Vivir: inhibición de la resorción ósea osteoclástica que, al modificar el microambiente de la médula ósea, la hace menos adecuada para el crecimiento de células tumorales; Actividad antiangiogénica y analgésica.
• In vitro: inhibición de la proliferación de osteoblastos, actividad citostática y proapoptótica directa sobre las células tumorales, efecto citostático sinérgico con otros fármacos anticancerosos, actividad inhibidora de la adhesión e invasión.
Resultados de ensayos clínicos en la prevención de eventos relacionados con el esqueleto en pacientes con tumores malignos avanzados que afectan al hueso
En el primer estudio aleatorizado, doble ciego y controlado con placebo, se comparó ácido zoledrónico 4 mg con placebo para la prevención de eventos relacionados con el esqueleto (ERE) en pacientes con cáncer de próstata con metástasis óseas. El ácido zoledrónico 4 mg redujo significativamente el porcentaje de pacientes con al menos un evento relacionado con el esqueleto (SRE), retrasó la mediana del tiempo hasta el primer SRE en> 5 meses y redujo la incidencia anual de eventos en> 5 meses. El análisis de eventos múltiples mostró una reducción del 36% en el riesgo de desarrollar SRE en el grupo de 4 mg de ácido zoledrónico en comparación con el placebo. Los pacientes tratados con 4 mg de ácido zoledrónico informaron menos aumento del dolor que los pacientes tratados con placebo, y la diferencia alcanzó significación en los meses 3, 9, 21 y 24. Menos pacientes tratados con 4 mg de ácido zoledrónico informaron fracturas patológicas. Los efectos del tratamiento fueron menos pronunciados en pacientes con lesiones blásticas. Los resultados de eficacia se muestran en la Tabla 2.
En un segundo estudio, que incluyó tumores sólidos distintos del cáncer de mama o el cáncer de próstata, 4 mg de ácido zoledrónico redujeron significativamente la proporción de pacientes con un SRE, retrasaron significativamente el tiempo medio hasta el primer SRE en> 2 meses y redujeron la tasa de morbilidad esquelética. El análisis de eventos múltiples mostró una reducción del 30,7% en el riesgo de desarrollar SRE en el grupo de 4 mg de ácido zoledrónico en comparación con el placebo. Los resultados de eficacia se muestran en la Tabla 3. Tabla 2: Resultados de eficacia (pacientes con cáncer de próstata tratados con terapia hormonal)
* Incluye fracturas vertebrales y no vertebrales
** Incluye todos los eventos esqueléticos, el número total y el tiempo hasta cada evento durante el estudio.
NR No logrado
NA No aplicable
Tabla 3: Resultados de eficacia (tumores sólidos distintos del cáncer de mama o de próstata)
* Incluye fracturas vertebrales y no vertebrales
** Incluye todos los eventos esqueléticos, el número total y el tiempo hasta cada evento durante el estudio.
NR No logrado
NA No aplicable
En un tercer estudio fase III, aleatorizado, doble ciego, se compararon 4 mg de ácido zoledrónico y 90 mg de pamidronato administrados cada 3 a 4 semanas en pacientes con mieloma múltiple o cáncer de mama con al menos una lesión ósea. Los resultados demostraron que el tratamiento con 4 mg de ácido zoledrónico produjo una eficacia comparable a la obtenida con pamidronato de 90 mg para la prevención de SRE. El análisis de múltiples eventos mostró una reducción significativa del 16% en el riesgo de desarrollar SRE en pacientes tratados con 4 mg de ácido zoledrónico en comparación con los tratados con pamidronato.Los resultados de eficacia se muestran en la Tabla 4.
Tabla 4: Resultados de eficacia (pacientes con cáncer de mama y mieloma múltiple)
* Incluye fracturas vertebrales y no vertebrales
** Incluye todos los eventos esqueléticos, el número total y el tiempo hasta cada evento durante el estudio.
NR No logrado
NA No aplicable
El ácido zoledrónico 4 mg también se estudió en 228 pacientes con metástasis óseas documentadas de cáncer de mama en un estudio doble ciego, aleatorizado y controlado con placebo para evaluar el efecto de 4 mg de ácido zoledrónico en el índice de morbilidad esquelética (SRE), calculado como el número total de eventos relacionados con el esqueleto (SRE) (excluyendo la hipercalcemia y corregidos por fractura previa), dividido por el tiempo total de riesgo. Los pacientes tomaron 4 mg de ácido zoledrónico o placebo cada cuatro semanas durante un año. Los pacientes se distribuyeron uniformemente entre los grupos de tratamiento con ácido zoledrónico y placebo.
La relación SRE (eventos / persona año) fue 0,628 para el ácido zoledrónico y 1,096 para el placebo. La proporción de pacientes con al menos un SRE (excluida la hipercalcemia) fue del 29,8% en el grupo de tratamiento con ácido zoledrónico en comparación con el 49,6% en el grupo de placebo ( p = 0,003). En el grupo de tratamiento con ácido zoledrónico, la mediana del tiempo hasta el inicio del primer SRE no se alcanzó durante la duración del estudio y se prolongó significativamente en comparación con el placebo (p = 0,007) Análisis de eventos múltiples (cociente de riesgos = 0,59, p = 0,019) mostró una reducción del 41% en el riesgo de desarrollar SRE en el grupo de 4 mg de ácido zoledrónico en comparación con el placebo.
En el grupo de tratamiento con ácido zoledrónico, hubo una mejora estadísticamente significativa en la puntuación del dolor (evaluada mediante el cuestionario del Inventario Breve de Dolor (BPI)) a partir de la semana 4 y para todas las evaluaciones posteriores realizadas durante el estudio en comparación con el placebo. Para el ácido zoledrónico, la puntuación de dolor estuvo constantemente por debajo del valor inicial y la reducción del dolor se asoció con una tendencia a la disminución de la puntuación de la terapia del dolor.
Resultados de ensayos clínicos en el tratamiento de la HIT
Los estudios clínicos en hipercalcemia neoplásica (HIT) han demostrado que el efecto del ácido zoledrónico se caracteriza por una disminución de calcio y de la excreción urinaria de calcio. En los estudios de búsqueda de dosis de fase I en pacientes con hipercalcemia neoplásica leve a moderada (HIT), las dosis efectivas probadas estuvieron aproximadamente en el rango de 1,2-2,5 mg.
Para verificar los efectos del ácido zoledrónico 4 mg en comparación con el pamidronato en una dosis de 90 mg, los resultados de dos ensayos clínicos "pivotales" multicéntricos en pacientes con HIT se combinaron para un "análisis predefinido. El ácido" zoledrónico en 8 mg , demostró una normalización más rápida de la concentración de calcio sérico el día 4 y, a 4 mg y 8 mg, el día 7. Se observaron las siguientes tasas de respuesta:
Tabla 5: Porcentaje de pacientes que mostraron una respuesta completa, (por día) en los estudios agrupados en TIH
La mediana de tiempo hasta la normalización del calcio fue de 4 días. La mediana del tiempo hasta la recaída (nuevo aumento de calcio corregido por albúmina sérica ≥ 2,9 mmol / l) osciló entre 30 y 40 días en pacientes tratados con ácido zoledrónico en comparación con 17 días en pacientes tratados con pamidronato de 90 mg (p: 0,001 para la dosis de 4 mg). y 0,007 para la dosis de 8 mg). No existen diferencias estadísticamente significativas entre las dos dosis diferentes de ácido zoledrónico.
En los ensayos clínicos, 69 pacientes que recayeron o fueron refractarios al tratamiento inicial (dosis de 4 mg, 8 mg de ácido zoledrónico o 90 mg de pamidronato) fueron tratados adicionalmente con 8 mg de ácido zoledrónico. La respuesta al tratamiento en estos pacientes fue aproximadamente del 52%. Como estos pacientes fueron tratados con la dosis de 8 mg solamente, no hay datos disponibles que permitan la comparación con la dosis de 4 mg.
En estudios clínicos en pacientes con hipercalcemia neoplásica (HIT), el perfil de seguridad global entre los tres grupos de tratamiento (ácido zoledrónico 4 mg y 8 mg y pamidronato 90 mg) fue similar en tipo y gravedad.
Población pediátrica
Resultados de estudios clínicos en el tratamiento de la osteogénesis imperfecta grave, en pacientes pediátricos de 1 a 17 años
Los efectos de la infusión intravenosa de ácido zoledrónico en el tratamiento de pacientes pediátricos (de 1 a 17 años) con osteogénesis imperfecta grave (tipos I, III y IV) se compararon con la infusión intravenosa de pamidronato, en un estudio internacional., Multicéntrico, aleatorizado , abierto con 74 y 76 pacientes en cada grupo de tratamiento, respectivamente. El período de tratamiento del estudio fue de 12 meses, precedido por un período de evaluación de 4 a 9 semanas durante el cual se administraron suplementos de vitamina D y calcio durante al menos 2 semanas. En el programa clínico, los pacientes de 1 a 3 años recibieron 0,025 mg / kg de ácido zoledrónico (hasta una dosis única máxima de 0,35 mg) cada 3 meses y los pacientes de 3 a 17 años recibieron 0,05 mg / kg de ácido zoledrónico (hasta una dosis única máxima de 0,83 mg) cada 3 meses. Se realizó un estudio de extensión para evaluar el perfil de seguridad renal y general a largo plazo del ácido zoledrónico administrado una o dos veces al año, durante 12 meses más, en niños que habían completado un año de tratamiento con ácido zoledrónico o pamidronato en el estudio principal. .
El criterio de valoración principal del estudio fue el cambio porcentual con respecto al valor inicial en la densidad mineral ósea (DMO) de la columna lumbar después de 12 meses de tratamiento. Los efectos esperados del tratamiento sobre la DMO fueron similares, pero el diseño del estudio no fue lo suficientemente sólido como para establecer la no- eficacia inferior del ácido zoledrónico. En particular, no hubo pruebas claras de eficacia sobre la incidencia de fracturas o el dolor. Se notificaron eventos adversos con fracturas de huesos largos de las extremidades inferiores en aproximadamente el 24% (fémur) y el 14% (tibia) de los pacientes con osteogénesis imperfecta grave tratados con ácido zoledrónico, frente al 12% y el 5% de los pacientes tratados con pamidronato, independientemente del tipo de enfermedad y la relación causal, pero la incidencia global de fracturas fue comparable entre los pacientes tratados con ácido zoledrónico y pamidronato: 43% (32/74) vs 41% (31/76). La interpretación del riesgo de fractura se dificulta por el hecho que las fracturas son eventos comunes en pacientes con osteogénesis imperfecta severa como parte del proceso de la enfermedad.
El tipo de reacciones adversas observadas en esta población fue similar a la observada previamente en adultos con neoplasias avanzadas que afectan al hueso (ver sección 4.8). Las reacciones adversas, clasificadas por orden de frecuencia, se presentan en la Tabla 6. Las reacciones adversas se clasifican según siguiente convención: muy frecuentes (≥1 / 10), frecuentes (≥1 / 100,
Tabla 6: Reacciones adversas observadas en pacientes pediátricos con osteogénesis imperfecta grave 1
1 Eventos adversos que ocurrieron con una frecuencia
En pacientes pediátricos con osteogénesis imperfecta grave, el ácido zoledrónico, en comparación con el pamidronato, parece estar asociado con riesgos más pronunciados de reacción de fase aguda, hipocalcemia y taquicardia inexplicable, pero esta diferencia se reduce después de infusiones posteriores.
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los estudios con ácido zoledrónico en todos los subgrupos de la población pediátrica para el tratamiento de la hipercalcemia neoplásica y la prevención de acontecimientos relacionados con el esqueleto en pacientes afectados. De tumores malignos avanzados que afectan al hueso (ver sección 4.2 para obtener información sobre uso pediátrico).
05.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos después de infusiones intravenosas únicas y repetidas de 5 y 15 minutos de 2, 4, 8 y 16 mg de ácido zoledrónico en 64 pacientes con metástasis óseas han mostrado los siguientes resultados independientemente de la dosis.
Después de iniciar la perfusión de ácido zoledrónico, las concentraciones plasmáticas de ácido zoledrónico aumentan rápidamente, alcanzando un máximo al final del período de perfusión, seguido de un rápido descenso a una concentración.
El ácido zoledrónico, administrado por vía intravenosa, se elimina según un proceso que se desarrolla en tres fases: rápida desaparición del fármaco de la circulación sistémica, con un curso bifásico, con una vida media plasmática de (t½α) 0,24 y (t½β) 1 , 87 horas, seguida de una fase de eliminación lenta con semivida de eliminación terminal de (t½γ) 146 horas No hay acumulación de ácido zoledrónico en plasma después de múltiples dosis administradas cada 28 días. El ácido zoledrónico no se metaboliza y se excreta inalterado a través de los riñones.Después de las primeras 24 horas, el 39 ± 16% de la dosis administrada está presente en la orina, mientras que el resto se une principalmente al tejido óseo. lentamente en la circulación sistémica y luego se elimina a través del riñón. El aclaramiento corporal es de 5,04 ± 2,5 l / h, independientemente de la dosis y no influido por el sexo, la edad, la raza y el peso corporal. El aumento del tiempo de infusión de 5 a 15 minutos produjo un 30 % de disminución de la concentración de ácido zoledrónico al final de la perfusión, pero no cambió el área bajo la curva (concentración plasmática frente al tiempo).
Al igual que con otros bifosfonatos, la variabilidad entre pacientes en los parámetros farmacocinéticos del ácido zoledrónico fue alta.
No se dispone de datos farmacocinéticos para el ácido zoledrónico en pacientes con hipercalcemia o en pacientes con insuficiencia hepática. In vitro, el ácido zoledrónico no inhibe las enzimas del citocromo P450 humano, no presenta biotransformación ni una cantidad de heces en estudios con animales, lo que respalda el hecho de que no existe un papel relevante de la función hepática en la farmacocinética del ácido zoledrónico.
El aclaramiento renal de ácido zoledrónico se correlacionó con el aclaramiento de creatinina, lo que representa un aclaramiento renal del 75 ± 33% del aclaramiento de creatinina, que promedió 84 ± 29 ml / min (rango 22 a 143 ml / min) en los 64 pacientes con cáncer estudiados. Los análisis de población mostraron que para un paciente con un aclaramiento de creatinina de 20 ml / min (insuficiencia renal grave) o 50 ml / min (insuficiencia renal moderada), el aclaramiento correspondiente previsto para el ácido zoledrónico debe corresponder al 37% o 72%, respectivamente, del de un paciente con un aclaramiento de creatinina de 84 ml / min. Solo se dispone de datos farmacocinéticos limitados en pacientes con insuficiencia renal grave (aclaramiento de creatinina
En un estudio in vitro, el ácido zoledrónico mostró una baja afinidad por los componentes celulares de la sangre humana, con una tasa de concentración plasmática media del 0,59% en un rango de 30 ng / mL a 5000 ng / mL. La unión a proteínas plasmáticas es baja, con la fracción libre que varía del 60% a 2 ng / mL al 77% a 2000 ng / mL de ácido zoledrónico.
Categorías especiales de pacientes
Pacientes pediátricos
Los datos farmacocinéticos limitados en niños con osteogénesis imperfecta grave sugieren que la farmacocinética del ácido zoledrónico en niños de 3 a 17 años es similar a la de los adultos cuando se considera un nivel de dosis similar (mg / kg), peso corporal, sexo y aclaramiento de creatinina. no parecen influir en la exposición sistémica del ácido zoledrónico.
05.3 Datos preclínicos sobre seguridad
Toxicidad aguda
La dosis máxima no letal para la administración intravenosa única fue de 10 mg / kg de peso corporal en el ratón y 0,6 mg / kg en la rata.
Toxicidad subcrónica y crónica
La tolerabilidad del ácido zoledrónico fue buena después de la administración subcutánea en la rata y la administración intravenosa en el perro a dosis de hasta 0.02 mg / kg / día durante 4 semanas. Administración subcutánea de 0.001 mg / kg / día en la rata y administración intravenosa de 0.005 mg / kg cada 2-3 días en perros hasta 52 semanas fue bien tolerado.
El hallazgo más frecuente en estudios de dosis repetidas es un aumento del tejido óseo esponjoso en las metáfisis de huesos largos en animales en desarrollo en aproximadamente todas las dosis, lo que refleja la actividad farmacológica del producto sobre la resorción ósea.
Se ha demostrado que los márgenes de seguridad para los efectos renales son pequeños en estudios a largo plazo en animales con dosis parenterales repetidas, pero los niveles acumulativos sin eventos adversos (NOAEL) por dosis única (1,6 mg / kg) y estudios de dosis múltiples hasta un mes ( 0,06-0,6 mg / kg / día) no han mostrado consecuencias renales a dosis equivalentes o superiores a la dosis terapéutica máxima en humanos. La administración repetida a largo plazo de grupos de dosis dentro de la dosis terapéutica máxima utilizada en humanos para el ácido zoledrónico produjo efectos tóxicos en otros órganos, incluidos el tracto gastrointestinal, el hígado, el bazo y los pulmones, así como en los lugares de inyección.
Estudios de toxicidad de la función reproductiva
Se demostró que el ácido zoledrónico es teratogénico en la rata después de la administración subcutánea de dosis ≥ 0,2 mg / kg. Se observó toxicidad materna en el conejo, aunque no se observaron efectos teratogénicos o fetotóxicos. A la dosis más baja probada en la rata (0,01 mg) / kg de peso corporal) se observó distocia.
Mutagénesis y carcinogénesis
En las pruebas de mutagenicidad realizadas, se demostró que el ácido zoledrónico no tiene efectos mutagénicos ni potencial carcinogénico.
06.0 INFORMACIÓN FARMACÉUTICA
06.1 Excipientes
Vial de polvo: manitol
Citrato de sodio
Vial de disolvente: agua para preparaciones inyectables.
06.2 Incompatibilidad
Para evitar posibles incompatibilidades, la solución reconstituida de Zometa debe diluirse con solución salina al 0,9% p / v o solución de glucosa al 5% p / v.
Este medicamento no debe mezclarse con soluciones para perfusión que contengan calcio u otros cationes divalentes como la solución de lactato de Ringer, y debe administrarse como una única solución intravenosa en una línea de perfusión separada.
06.3 Período de validez
3 años.
Después de la reconstitución y dilución: Desde un punto de vista microbiológico, la solución para perfusión reconstituida y diluida debe usarse inmediatamente. Si no se usa inmediatamente, los tiempos de almacenamiento en uso y las condiciones antes de su uso son responsabilidad del usuario y normalmente no deben exceder las 24 horas a 2 C - 8 C. La solución refrigerada debe llevarse a temperatura ambiente antes de su administración.
06.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
Para las condiciones de conservación de la solución reconstituida para perfusión, ver sección 6.3.
06.5 Naturaleza del envase primario y contenido del envase.
Vial de polvo: Viales de vidrio incoloro de 6 ml, vidrio de grado hidrolítico tipo I (Ph. Eur.).
Vial de disolvente: Vial de vidrio incoloro de 5 ml.
Envases individuales que contienen respectivamente 1 o 4 viales y 1 o 4 ampollas de agua para preparaciones inyectables.
Envases múltiples que contienen 10 (10 envases de 1 + 1) viales y ampollas de agua para preparaciones inyectables.
Es posible que no se comercialicen todos los tamaños de envases.
06.6 Instrucciones de uso y manipulación
El polvo debe reconstituirse previamente en 5 ml de agua para preparaciones inyectables utilizando el vial especial incluido en el envase del producto La disolución del polvo debe ser completa antes de tomar la solución para su posterior dilución. La cantidad necesaria de solución reconstituida debe diluirse adicionalmente en 100 ml de solución para perfusión sin calcio (solución salina al 0,9% p / v o solución de glucosa al 5% p / v).
En la sección 4.2 se proporciona información adicional sobre la manipulación de Zometa, incluida una guía sobre la preparación de dosis reducidas.
Se deben seguir técnicas asépticas durante la preparación de la perfusión Para un solo uso.
Solo se debe utilizar la solución transparente, libre de partículas visibles e incolora.
Se debe advertir a los profesionales de la salud que no desechen el Zometa no utilizado por el sistema de recolección de residuos domésticos.
Los medicamentos no utilizados y los desechos derivados de este medicamento deben eliminarse de acuerdo con las regulaciones locales.
07.0 TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
Parque empresarial Frimley
Camberley GU16 7SR
Reino Unido
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU / 1/01/176 / 001-003
035263033
035263019
035263021
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 20.03.2001
Fecha de la última renovación: 20.03.2006
10.0 FECHA DE REVISIÓN DEL TEXTO
D.CCE julio de 2015