A menudo, una consecuencia de ciertos tipos de medicamentos, el síndrome de Stevens-Johnson a veces puede ser causado por infecciones u otros factores.
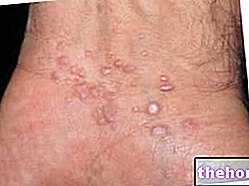
La descamación de la piel es uno de los síntomas característicos del síndrome en cuestión; Los pacientes que se someten a su desarrollo suelen ser hospitalizados y se someten a los cuidados y tratamientos de apoyo necesarios. Si el síndrome es causado por drogas, su ingesta debe detenerse de inmediato.
Si se diagnostica con prontitud, el pronóstico es generalmente bueno.
y mucosas (necrólisis) cuyas consecuencias pueden ser sumamente graves.
La incidencia anual del síndrome de Stevens-Johnson es de aproximadamente 1-5 / 1,000,000, por lo tanto, afortunadamente, esta es una reacción muy rara.
Varias fuentes consideran el síndrome de Stevens-Johnson como una variante limitada de necrólisis epidérmica tóxica (o síndrome de Lyell, un tipo particular de eritema polimórfico), caracterizado por una sintomatología similar pero más extensa y severa que la del síndrome de Stevens-Johnson.
Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica: ¿cuáles son las diferencias?
Los cuadros sintomáticos del síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica son muy similares entre sí. Ambos se caracterizan por la destrucción y posterior descamación de la piel que, sin embargo, en el síndrome objeto del artículo afecta áreas corporales limitadas (menos del 10% de la superficie corporal total); mientras que en la necrólisis epidérmica tóxica afecta áreas más grandes de la piel (más del 30% de la superficie corporal).
La afectación del 15% al 30% de la superficie cutánea se considera, por otro lado, como una superposición entre el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica.
(como, por ejemplo, cotrimoxazol y sulfasalazina);Otras causas
Otras posibles causas responsables de la aparición del síndrome de Stevens-Johnson son:
- Infecciones de naturaleza viral o bacteriana (en particular, sostenidas por Micoplasma pneumoniae);
- La administración de algunos tipos de vacunas;
- Enfermedad de injerto contra huésped (o EICH).
¿Qué personas corren el riesgo de desarrollar el síndrome de Stevens-Johnson?
El riesgo de desarrollar el síndrome de Stevens-Johnson es mayor:
- En pacientes VIH positivos y con SIDA;
- En pacientes con deterioro del sistema inmunológico (por ejemplo, debido a medicamentos);
- En pacientes con infecciones causadas por Pneumocystis jirovecii;
- En pacientes con lupus eritematoso sistémico;
- En pacientes que padecen enfermedades reumáticas crónicas;
- En pacientes con antecedentes de síndrome de Stevens-Johnson.
Finalmente, también se ha adelantado la hipótesis de la existencia de una cierta predisposición genética al desarrollo del síndrome de Stevens-Johnson.
;
En esta etapa, muchos pacientes también pueden experimentar una sensación de ardor inexplicable y / o dolor en la piel.
En caso de que el síndrome de Stevens-Johnson sea causado por medicación, los síntomas prodrómicos anteriores aparecen dentro de 1-3 semanas después de iniciar la terapia, sin embargo, los síntomas cutáneos y mucosos descritos a continuación, aparecen después de 4-6 semanas desde el inicio de la misma.
Síntomas que afectan la piel y las membranas mucosas.
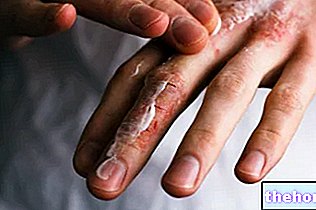
Después del inicio de los síntomas prodrómicos antes mencionados, surgen síntomas que afectan la piel y las membranas mucosas. Comienzan con una "erupción plana y roja que generalmente comienza en la cara, el cuello y el tronco y luego se extiende al resto del cuerpo. En el caso específico del síndrome de Stevens-Johnson, esta erupción afecta menos del 10% de la superficie". corpóreo.
La aparición de la erupción es seguida por la formación de ampollas que tienden a exfoliarse en 1-3 días. Las ampollas también pueden aparecer en los genitales, manos, pies; ocurren en las membranas mucosas (por ejemplo, en la boca, garganta, etc.) e incluso puede afectar a los epitelios internos, como los de las vías respiratorias, el tracto urinario, etc. Los ojos también se ven afectados generalmente por la formación de ampollas y costras: están hinchados, enrojecidos y dolorosos.
Además de la descamación marcada de la piel, también se puede encontrar la pérdida de uñas y cabello.
Claramente, en tal condición, el paciente percibe un dolor considerable que está asociado con una "hinchazón igualmente notable. Además, dependiendo de las áreas donde se forman las ampollas y donde ocurre la descamación, el paciente puede desarrollar dificultades para respirar, dificultad para orinar, dificultad para mantener su ojos abiertos, dificultad para tragar, hablar, comer e incluso beber.
Complicaciones del síndrome de Stevens-Johnson
Las complicaciones del síndrome de Stevens-Johnson ocurren principalmente debido a la necrosis y posterior descamación de la piel y las membranas mucosas. Al carecer de la función de barrera que normalmente ejerce la piel, de hecho, uno puede encontrar:
- Pérdida masiva de electrolitos y líquidos;
- Contracción de varios tipos de infecciones (bacterianas, virales, fúngicas, etc.) que también pueden provocar sepsis.
Otra complicación grave consiste en la "aparición" de una insuficiencia que afecta a varios órganos (insuficiencia multiorgánica).
piel que resulta en un examen histológico, aunque éste no es un procedimiento que se realice con frecuencia.
Desafortunadamente, el diagnóstico temprano, cuando el síndrome aún está en su infancia y se manifiesta con síntomas prodrómicos, no siempre es posible.De hecho, dado que estos síntomas son bastante inespecíficos, se podría encontrar una "evaluación errónea", con el consiguiente retraso en la identificación de la causa real del malestar del paciente.
¿Con qué enfermedades no se debe confundir el síndrome de Stevens-Johnson?
Las manifestaciones y síntomas inducidos por el síndrome de Stevens-Johnson pueden ser similares a los inducidos por otras enfermedades que afectan la piel y las mucosas, con las que, sin embargo, no debe confundirse. Más en detalle, el diagnóstico diferencial debe situarse en relación con:
- Dell "eritema polimórfico menor y mayor;
- De necrólisis epidérmica tóxica o síndrome de Lyell, si lo prefiere;
- Síndrome de shock tóxico;
- De dermatitis exfoliativa;
- Pénfigo.
Si el síndrome es causado por la ingesta de medicamentos, el tratamiento con los medicamentos debe suspenderse inmediatamente.
Desafortunadamente, aunque existen algunos medicamentos que se pueden administrar para tratar de detener la progresión del síndrome, no existe una cura real para el síndrome de Stevens-Johnson. En cualquier caso, los pacientes hospitalizados por el síndrome en cuestión deben recibir tratamientos de soporte adecuados.
Terapia de apoyo
Los cuidados de apoyo son esenciales para garantizar la supervivencia del paciente. Puede variar según las condiciones de este último.
- Los líquidos y electrolitos perdidos deberán administrarse por vía parenteral. Lo mismo se aplica a la nutrición si el paciente no puede proporcionarla de forma independiente debido a las lesiones reportadas en la piel y membranas mucosas de la boca, garganta, etc.
- Los pacientes con afectación ocular deberán someterse a visitas a especialistas y a tratamientos específicos para prevenir, o al menos limitar, el daño inducido por el síndrome.
- Las lesiones cutáneas típicas del síndrome de Stevens-Johnson deben tratarse a diario y tratarse de la misma forma que las quemaduras.
- En presencia de infecciones secundarias, es necesario proceder con el tratamiento de estas últimas, estableciendo terapias adecuadas contra el patógeno desencadenante (utilizando, por ejemplo, antibióticos, siempre que no se sepa que induzcan el síndrome en cuestión, medicamentos antifúngicos, etc.).
Medicamentos para reducir la duración del síndrome de Stevens-Johnson
En un intento por frenar el síndrome de Stevens-Johnson o en cualquier caso reducir su duración, es posible recurrir al uso de fármacos, como:
- Ciclosporina, que se administra para inhibir la acción de las células T.
- Corticosteroides de uso sistémico, con el fin de amortiguar la acción del sistema inmunológico, pero al mismo tiempo útiles para contrarrestar el dolor.
- Inmunoglobulinas intravenosas (IgEV) en dosis altas; administrado temprano. Deben detener la actividad de los anticuerpos y bloquear la acción del ligando del receptor FAS.
Sin embargo, el uso de estos medicamentos es objeto de opiniones divergentes entre los médicos.
El uso de corticoides, de hecho, se correlaciona con un aumento de la mortalidad y puede favorecer la aparición de infecciones o enmascarar una posible sepsis; aunque dichos fármacos han demostrado cierta eficacia para mejorar las lesiones oculares.
Asimismo, aunque la administración de IgEV permite obtener buenos resultados iniciales, según ensayos clínicos y análisis retrospectivos, los resultados finales obtenidos son contradictorios. De hecho, a partir de estos experimentos y análisis, resultó que la IgEV no solo puede ser ineficaz, sino que puede estar potencialmente relacionada con un aumento de la mortalidad.
Otros tratamientos
En algunos casos, también se puede realizar plasmaféresis para eliminar residuos de metabolitos reactivos de fármacos y anticuerpos que podrían ser la causa desencadenante del síndrome de Stevens-Johnson.
En cualquier caso, el médico establecerá el tratamiento más adecuado para cada paciente en función del estado del mismo y en función de la etapa en la que se encuentre el síndrome de Stevens-Johnson en el momento de su diagnóstico.