Ingredientes activos: Fondaparinux (fondaparinux sódico)
Arixtra 1,5 mg / 0,3 ml solución inyectable.
Los prospectos de Arixtra están disponibles para los siguientes tamaños de envase:- Arixtra 1,5 mg / 0,3 ml solución inyectable.
- Arixtra 2,5 mg / 0,5 ml solución inyectable.
- Arixtra 5 mg / 0,4 ml solución inyectable, Arixtra 7,5 mg / 0,6 ml solución inyectable, Arixtra 10 mg / 0,8 ml solución inyectable
¿Por qué se usa Arixtra? ¿Para qué sirve?
Arixtra es un medicamento que ayuda a prevenir la formación de coágulos de sangre en los vasos sanguíneos (agente antitrombótico).
Arixtra contiene una sustancia llamada fondaparinux sódico. Actúa inhibiendo la actividad del factor de coagulación Xa ("diez-A") en la sangre, evitando así la formación de coágulos de sangre (trombosis) en los vasos sanguíneos.
Arixtra se utiliza para:
- evitar que se formen coágulos de sangre en los vasos sanguíneos de las piernas o los pulmones después de una cirugía ortopédica (como una cirugía de cadera o rodilla) o después de una cirugía abdominal;
- prevenir la formación de coágulos de sangre durante e inmediatamente después de un período de movilidad limitada debido a una enfermedad aguda;
- el tratamiento de los coágulos de sangre en los vasos sanguíneos superficiales de las piernas (trombosis de las venas superficiales).
Contraindicaciones Cuándo no se debe usar Arixtra
No use Arixtra:
- si es alérgico al fondaparinux sódico oa cualquiera de los demás componentes de este medicamento
- si tiene sangrado abundante;
- si tiene una "infección bacteriana del corazón;
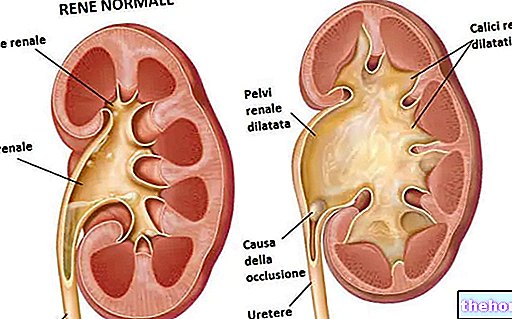
- si tiene una enfermedad renal muy grave.
Informe a su médico si cree que se encuentra en alguna de estas situaciones. En ese caso, no debe utilizar Arixtra.
Precauciones de uso Qué necesita saber antes de tomar Arixtra
Tenga especial cuidado con Arixtra:
Consulte a su médico o farmacéutico antes de tomar Arixtra:
- si tiene riesgo de sangrado incontrolado (hemorragias) que incluyen: úlcera de estómago enfermedad hemorrágica hemorragia cerebral reciente (hemorragia intracraneal) cirugía cerebral, de la columna vertebral o ocular reciente
- si tiene una enfermedad grave del hígado
- si tiene una enfermedad del riñón
- si tienes 75 años o más
- si pesa menos de 50 kg.
Informe a su médico si cree que se encuentra en alguna de estas situaciones.
Niños y adolescentes
Arixtra no se ha probado para su uso en niños y adolescentes menores de 17 años.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Arixtra?
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto también incluye los comprados sin receta. Algunos otros medicamentos pueden afectar la forma en que actúa Arixtra o pueden verse afectados por Arixtra.
Advertencias Es importante saber que:
Embarazo y lactancia
Arixtra no se debe recetar a mujeres embarazadas a menos que sea específicamente necesario. No se recomienda amamantar mientras toma Arixtra. Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Arixtra contiene sodio
Cada dosis de este medicamento contiene menos de 23 mg de sodio y, por tanto, es esencialmente libre de sodio.
La jeringa Arixtra contiene látex
La cubierta de la aguja de la jeringa contiene látex que tiene el potencial de causar reacciones alérgicas en personas sensibles al látex.
- Informe a su médico si tiene alergia al látex antes de recibir tratamiento con Arixtra.
Dosis, método y momento de administración Cómo utilizar Arixtra: Posología
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte a su médico o farmacéutico.
La dosis recomendada es de 2,5 mg una vez al día, que se inyecta aproximadamente a la misma hora todos los días.
Si tiene una enfermedad renal, la dosis se puede reducir a 1,5 mg una vez al día.
Cómo se administra Arixtra
- Arixtra se administra mediante inyección debajo de la piel (por vía subcutánea) en un pliegue cutáneo en la zona abdominal inferior.Las jeringas están precargadas con la dosis exacta necesaria. Las jeringas de dosificación de 2,5 mg y 1,5 mg son diferentes. Para las "Instrucciones de uso" punto por punto, consulte el final de la hoja.
- No inyecte Arixtra en el músculo.
¿Cuánto tiempo debe tomarse Arixtra?
Debe continuar el tratamiento con Arixtra durante el tiempo que le indique su médico, ya que Arixtra previene el desarrollo de enfermedades graves.
Sobredosis Qué hacer si ha tomado demasiado Arixtra
Si se inyecta demasiado Arixtra
Póngase en contacto con su médico o farmacéutico lo antes posible para que le aconseje, ya que esto aumenta el riesgo de hemorragia.
Si olvidó tomar Arixtra
- Administre la dosis tan pronto como se acuerde. No inyecte una dosis doble para compensar las dosis olvidadas.
- Si no está seguro de qué hacer, consulte a su médico o farmacéutico.
No deje de usar Arixtra sin consejo médico.
Si interrumpe el tratamiento antes de lo prescrito por su médico, corre el riesgo de desarrollar un coágulo de sangre en una vena de las piernas o en los pulmones. Póngase en contacto con su médico o farmacéutico antes de interrumpir el tratamiento.
Si tiene más preguntas sobre el uso de este medicamento, consulte a su médico o farmacéutico.
Efectos secundarios ¿Cuáles son los efectos secundarios de Arixtra?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Condiciones para las que es necesario pedir ayuda.
Reacciones alérgicas graves (anafilaxia): son muy raras en personas que toman Arixtra (hasta 1 de cada 10.000 personas). Los síntomas incluyen:
- hinchazón, a veces de la cara o la boca (angioedema), que causa dificultad para tragar o respirar
- colapso.
Comuníquese con su médico de inmediato si experimenta tales síntomas. Deje de tomar Arixtra.
Efectos secundarios comunes
Pueden afectar a más de una de cada 100 personas tratadas con Arixtra:
- sangrado (por ejemplo, en el lugar de la operación, de una úlcera de estómago preexistente, de la nariz, de las encías),
- anemia (reducción del número de glóbulos rojos).
Efectos secundarios poco frecuentes
Pueden afectar hasta una de cada 100 personas tratadas con Arixtra:
- hematomas o hinchazón (edema)
- sentirse o estar enfermo (náuseas o vómitos)
- Dolor de pecho
- dificultad para respirar
- enrojecimiento o picazón
- líquido que rezuma de la herida de la cirugía
- fiebre
- disminución o aumento del número de plaquetas (células sanguíneas necesarias para la coagulación)
- aumento de algunas sustancias (enzimas) producidas por el hígado
Efectos secundarios raros
Pueden afectar hasta 1 de cada 1.000 personas tratadas con Arixtra:
- reacciones alérgicas (que incluyen picazón, hinchazón, erupción cutánea)
- hemorragia cerebral interna o abdominal
- ansiedad o confusión
- dolor de cabeza
- desmayos o mareos, presión arterial baja
- somnolencia o cansancio
- Sofocos
- tos
- dolor de pierna o dolor de estómago
- diarrea o estreñimiento
- indigestión
- Infección en la herida
- aumento de la bilirrubina (una sustancia producida por el hígado) en la sangre
- reducción de potasio en sangre.
Notificación de efectos secundarios
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluidos los posibles efectos adversos que no aparecen en este prospecto. También puede notificar los efectos secundarios directamente a través del sistema de notificación nacional que figura en el Apéndice V. Al notificar los efectos secundarios, puede ayudar a proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
- Mantenga este medicamento fuera de la vista y del alcance de los niños.
- Conservar por debajo de 25 ° C. No congelar.
- Arixtra no debe conservarse en nevera.
No use este medicamento:
- después de la fecha de caducidad que se indica en la etiqueta y la caja
- si nota la presencia de partículas en la solución o si la solución tiene un color anormal
- si nota que la jeringa está dañada
- si ha abierto una jeringa y no la use inmediatamente.
Eliminación de jeringas:
No arroje ningún medicamento o jeringa a los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que no necesita, esto ayudará a proteger el medio ambiente.
Contenido del envase e información adicional
Qué contiene Arixtra
- El principio activo es 1,5 mg de fondaparinux sódico en 0,3 ml de solución inyectable.
- Los demás componentes son cloruro de sodio, agua para preparaciones inyectables y ácido clorhídrico y / o hidróxido de sodio para ajustar el pH.
Arixtra no contiene ningún producto de origen animal.
Descripción del aspecto de Arixtra y contenido del envase
Arixtra es una solución inyectable transparente e incolora. Se suministra con una jeringa precargada desechable, completa con un sistema de protección que ha sido diseñado para proteger contra pinchazos accidentales con la aguja después de su uso. Está disponible en envases de 2, 7, 10 y 20 jeringas precargadas (no todas tamaños de envases pueden comercializarse).
PUNTO A PUNTO UTILIZANDO LA GUÍA ARIXTRA
Instrucciones de uso
Estas instrucciones son válidas para ambos tipos de jeringas (sistema de protección de aguja automático y manual)
Cuando las instrucciones para cada jeringa sean diferentes, esto se indicará claramente.
1. Lávese bien las manos con agua y jabón y luego séquelas con una toalla.
2. Saque la jeringa del estuche y verifique que:
- la fecha de vencimiento no ha pasado
- la solución es transparente e incolora y no contiene partículas
- la jeringa no ha sido abierta o dañada
3. Siéntese o acuéstese en una posición cómoda.
Elija un punto en la zona abdominal inferior, al menos 5 cm por debajo del ombligo
Cambie los lados izquierdo y derecho de la zona inferior del abdomen con cada inyección, lo que ayudará a reducir las molestias en el lugar de la inyección.
Si no es posible la inyección en la zona abdominal inferior, consulte con su médico o enfermera.
4. Limpie el lugar de la inyección con un hisopo con alcohol.
5. Quite la cubierta de la aguja girándola primero y luego tirando de ella hacia afuera del cuerpo de la jeringa. Quite la tapa.
Nota IMPORTANTE
- No toque la aguja y asegúrese de que no entre en contacto con otras superficies antes de inyectarse.
- La presencia de una pequeña burbuja de aire en la jeringa es normal. No intente eliminar las pequeñas burbujas de aire antes de inyectarse para asegurarse de no perder ningún producto.
6. Pellizque ligeramente el área de piel desinfectada para formar un pliegue. Sostenga el pliegue entre el pulgar y el índice durante la inyección.
7. Sostenga la jeringa firmemente entre sus dedos.
Inserte perpendicularmente (en un ángulo de 90 °) toda la longitud de la aguja en el pliegue de la piel.
8. Inyecte TODO el contenido de la jeringa empujando el émbolo hacia abajo tanto como sea posible.
Jeringa con sistema automático
9. Suelte el émbolo y la aguja se retirará automáticamente de la piel hacia la funda de seguridad, donde permanecerá permanentemente cerrada.
Jeringa con sistema manual
9. Después de la inyección, sujete la jeringa con una mano mientras sujeta la funda de seguridad, use la otra mano para sujetar el asa y tire firmemente hacia atrás. Esto desbloquea la funda. Deslice la funda a través del cuerpo de la jeringa hasta que encaje en su lugar. sobre la aguja
No deseche la jeringa usada con la basura doméstica. Deseche la jeringa usada siguiendo las instrucciones que le haya dado su médico o farmacéutico.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO
ARIXTRA 1,5 MG / 0,3 ML
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada jeringa precargada (0,3 ml) contiene 1,5 mg de fondaparinux sódico.
Excipientes con efectos conocidos: Contiene menos de 1 mmol de sodio (23 mg) por dosis y, por lo tanto, esencialmente no contiene sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA
Solución inyectable.
La solución es un líquido transparente e incoloro.
04.0 INFORMACIÓN CLÍNICA
04.1 Indicaciones terapéuticas
Prevención de episodios tromboembólicos venosos (TEV) en adultos sometidos a cirugía ortopédica mayor de miembros inferiores, como fractura de cadera, cirugía mayor de rodilla o artroplastia de cadera.
Prevención de episodios tromboembólicos venosos (TEV) en adultos sometidos a cirugía abdominal considerados de alto riesgo de complicaciones tromboembólicas, como pacientes sometidos a cirugía abdominal por cáncer (ver sección 5.1).
Prevención de episodios tromboembólicos venosos (TEV) en adultos médicamente relevantes que se considera que tienen un alto riesgo de TEV y que están inmovilizados debido a una afección aguda como insuficiencia cardíaca y / o enfermedad respiratoria aguda y / o enfermedad o infección inflamatoria aguda.
Tratamiento de adultos con trombosis venosa superficial aguda espontánea sintomática de las extremidades inferiores en ausencia de trombosis venosa profunda concomitante (ver secciones 4.2 y 5.1).
04.2 Posología y forma de administración
Dosis
Pacientes sometidos a cirugía mayor ortopédica o abdominal
La dosis recomendada de fondaparinux es de 2,5 mg administrados una vez al día después de la cirugía mediante inyección subcutánea.
La dosis inicial debe administrarse 6 horas después del final de la cirugía una vez que se haya asegurado la hemostasia.
El tratamiento debe continuarse hasta que disminuya el riesgo de tromboembolismo venoso, generalmente hasta que el paciente reanude la marcha, al menos 5-9 días después de la cirugía.La experiencia muestra que en pacientes sometidos a cirugía por fractura de cadera el riesgo de TEV persiste más allá de los 9 días posteriores a la cirugía. En estos pacientes, se debe considerar el uso de profilaxis prolongada con fondaparinux hasta por 24 días adicionales (ver sección 5.1).
Pacientes médicamente relevantes que tienen un alto riesgo de complicaciones tromboembólicas según una evaluación de riesgo individual
La dosis recomendada de fondaparinux es de 2,5 mg una vez al día administrada por inyección subcutánea. El tratamiento que dura de 6 a 14 días se ha estudiado clínicamente en pacientes médicamente relevantes (ver sección 5.1).
Tratamiento de la trombosis venosa superficial.
La dosis recomendada de fondaparinux es de 2,5 mg al día, administrada por inyección subcutánea. Los pacientes aptos para el tratamiento con fondaparinux 2,5 mg deben presentar trombosis venosa superficial espontánea, aguda, sintomática y aislada de los miembros inferiores, de al menos 5 cm de longitud y documentada mediante ecografía u otras exploraciones físicas. El tratamiento debe iniciarse lo antes posible inmediatamente después del diagnóstico y después de la exclusión de trombosis venosa profunda (TVP) o trombosis venosa superficial concomitantes dentro de los 3 cm de la unión safeno-femoral. El tratamiento debe continuarse durante un mínimo de 30 días y hasta un máximo de 45 días en pacientes con alto riesgo de complicaciones tromboembólicas (ver secciones 4.4 y 5.1).
Se debe recomendar a los pacientes que se autoinyecten el producto cuando, a juicio del médico, estén dispuestos y puedan hacerlo. Los médicos deben proporcionar instrucciones claras para la autoinyección.
• Pacientes que necesitan someterse a cirugía u otros procedimientos invasivos.
En pacientes con trombosis venosa superficial que vayan a ser sometidos a cirugía u otros procedimientos invasivos, no se debe administrar fondaparinux, cuando sea posible, durante las 24 horas previas a la cirugía. El tratamiento con fondaparinux puede reiniciarse al menos 6 horas después de la cirugía. logrado.
Categorías especiales de pacientes
En pacientes sometidos a cirugía, el tiempo de administración de la primera inyección de fondaparinux requiere un estricto cumplimiento en pacientes ≥ 75 años y / o insuficiencia renal peso con un aclaramiento de creatinina entre 20 y 50 ml / min.
La primera administración de fondaparinux no debe administrarse antes de las 6 horas posteriores al final de la cirugía La inyección no debe administrarse sin que se haya establecido la hemostasia (ver sección 4.4).
Insuficiencia renal -
• Prevención de TEV - Fondaparinux no debe utilizarse en pacientes con aclaramiento de creatinina de 50 ml / min).
• Tratamiento de la trombosis venosa superficial. - Fondaparinux no debe utilizarse en pacientes con aclaramiento de creatinina de 50 ml / min). No se ha estudiado la seguridad y eficacia de 1,5 mg (ver sección 4.4).
Insuficiencia hepática
• Prevención de TEV - No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. En pacientes con insuficiencia hepática grave, fondaparinux debe utilizarse con precaución ya que no se ha estudiado en este grupo de pacientes (ver secciones 4.4 y 5.2).
• Tratamiento de la trombosis venosa superficial - No se ha estudiado la seguridad y eficacia de fondaparinux en pacientes con insuficiencia hepática grave, por lo que no se recomienda el uso de fondaparinux en estos pacientes (ver sección 4.4).
Población pediátrica - No se recomienda el uso de fondaparinux en niños menores de 17 años debido a la falta de datos sobre seguridad y eficacia.
Peso corporal bajo
• Prevención de TEV - Pacientes con peso corporal sangrante. La eliminación de fondaparinux disminuye con el peso, por lo que fondaparinux debe utilizarse con precaución en estos pacientes (ver sección 4.4).
• Tratamiento de la trombosis venosa superficial - No se ha estudiado la seguridad y eficacia de fondaparinux en pacientes que pesen menos de 50 kg, por lo que no se recomienda el uso de fondaparinux en estos pacientes (ver sección 4.4).
Método de administración
Fondaparinux se administra mediante inyección subcutánea profunda, con el paciente en decúbito supino. El lugar de la inyección debe alternar entre el lado anterolateral izquierdo y derecho y entre el lado posterolateral izquierdo y derecho de la pared abdominal. Para evitar la pérdida de medicamento al utilizar la jeringa precargada, no expulse burbujas de aire de la jeringa antes de la inyección. Toda la longitud de la aguja debe insertarse perpendicularmente en un pliegue de piel sostenido entre el pulgar y el índice; el pliegue cutáneo debe mantenerse mientras dure la inyección.
Para obtener más instrucciones de uso y eliminación, consulte la sección 6.6.
04.3 Contraindicaciones
- Hipersensibilidad conocida al principio activo oa alguno de los excipientes incluidos en la sección 6.1.
- sangrado en curso, clínicamente significativo
- endocarditis bacteriana aguda
- insuficiencia renal grave definida como aclaramiento de creatinina
04.4 Advertencias especiales y precauciones de uso apropiadas
Fondaparinux está destinado únicamente para uso subcutáneo, no debe administrarse por vía intramuscular.
Hemorragias
Fondaparinux debe usarse con precaución en pacientes que tienen un mayor riesgo de hemorragia, como aquellos con trastornos hemorrágicos congénitos o adquiridos (por ejemplo, recuento de plaquetas 3), enfermedad gastrointestinal ulcerativa activa y hemorragia cerebral, espinal o intracraneal reciente o poco después. en grupos especiales de pacientes como se indica a continuación.
• Para la prevención de TEV - Los agentes que pueden aumentar el riesgo de hemorragia no deben administrarse concomitantemente con fondaparinux. Tales sustancias incluyen desirudina, agentes fibrinolíticos, antagonistas del receptor GP IIb / IIIa, heparina, heparinoides o heparina de bajo peso molecular (HBPM). Si es necesario, se debe administrar un tratamiento concomitante con antagonistas de la vitamina K de acuerdo con las instrucciones de la sección 4.5. Otros fármacos antiplaquetarios (ácido acetilsalicílico, dipiridamol, sulfinpirazona, ticlopidina o clopidogrel) y AINE deben utilizarse con precaución. Si la coadministración es esencial, se requiere una estrecha vigilancia.
• Para el tratamiento de la trombosis venosa superficial. - Fondaparinux debe usarse con precaución en pacientes que reciben medicamentos concomitantes que aumentan el riesgo de hemorragia.
Pacientes con trombosis venosa superficial
La presencia de trombosis venosa superficial a una distancia superior a 3 cm de la unión safeno-femoral debe confirmarse antes de iniciar el tratamiento con fondaparinux y debe descartarse la presencia de TVP mediante ecografía de compresión (CUS) u otros métodos objetivos. No existen datos sobre el uso de fondaparinux 2,5 mg en pacientes con trombosis venosa superficial asociada con TVP concomitante o con trombosis venosa superficial dentro de los 3 cm de la unión safeno-femoral (ver secciones 4.2 y 5.1).
No se ha estudiado la seguridad y eficacia de fondaparinux 2,5 mg en los siguientes grupos: pacientes con trombosis venosa superficial después del tratamiento esclerosante o como consecuencia de una vía intravenosa, pacientes con antecedentes de trombosis venosa superficial en los 3 meses anteriores, pacientes con antecedentes de enfermedad tromboembólica venosa en los 6 meses anteriores o pacientes con un tumor activo (ver secciones 4.2 y 5.1).
Anestesia espinal / epidural
En pacientes sometidos a cirugía ortopédica mayor, con el uso concomitante de fondaparinux y anestesia espinal / epidural o punción espinal, no se puede excluir la aparición de hematomas epidurales o espinales que pueden resultar en parálisis prolongada o permanente. El riesgo de estos eventos raros puede aumentar con el uso postoperatorio de catéteres epidurales permanentes o con el uso concomitante de otros fármacos que actúan sobre la hemostasia.
Pacientes de edad avanzada
La población de edad avanzada tiene un mayor riesgo de hemorragia. Dado que la función renal generalmente disminuye con la edad, los pacientes de edad avanzada pueden mostrar una eliminación reducida y una mayor exposición a fondaparinux (ver sección 5.2). Fondaparinux debe usarse con precaución en pacientes de edad avanzada (ver sección 4.2).
Peso corporal bajo
• Prevención de TEV - Pacientes con peso corporal
• Tratamiento de la trombosis venosa superficial - No se dispone de datos clínicos sobre el uso de fondaparinux para el tratamiento de la trombosis venosa superficial en pacientes que pesen menos de 50 kg, por lo que no se recomienda fondaparinux para el tratamiento de la trombosis venosa superficial en estos pacientes (ver sección 4.2).
Insuficiencia renal
• Prevención de TEV - Se sabe que el fondaparinux se excreta principalmente por los riñones. Pacientes con aclaramiento de creatinina
• Tratamiento de la trombosis venosa superficial. - Fondaparinux no debe utilizarse en pacientes con aclaramiento de creatinina.
Insuficiencia hepática grave
• Prevención de TEV - No es necesario ajustar la dosis de fondaparinux. Sin embargo, el uso de fondaparinux en pacientes con insuficiencia hepática grave debe considerarse con precaución debido a un mayor riesgo de hemorragia debido a la deficiencia de factores de coagulación en pacientes con insuficiencia hepática grave (ver sección 4.2).
• Tratamiento de la trombosis venosa superficial. - No se dispone de datos clínicos sobre el uso de fondaparinux para el tratamiento de la trombosis venosa superficial en pacientes con
insuficiencia hepática grave.En consecuencia, no se recomienda fondaparinux para el tratamiento de la trombosis venosa superficial en estos pacientes (ver sección 4.2).
Pacientes con trombocitopenia inducida por heparina
Fondaparinux debe usarse con precaución en pacientes con antecedentes de trombocitopenia inducida por heparina (TIH). No se ha estudiado formalmente la eficacia y seguridad de fondaparinux en pacientes con TIH tipo II. Fondaparinux no se une al factor de coagulación 4 y no reacciona de forma cruzada con el plasma de pacientes con TIH tipo II. Se han recibido raras notificaciones espontáneas de TIH. en pacientes tratados con fondaparinux Hasta la fecha, no se ha establecido una asociación causal entre el tratamiento con fondaparinux y la aparición de TIH.
Alergia al latex
La tapa de la aguja de la jeringa precargada contiene látex de caucho natural seco que puede provocar reacciones alérgicas en personas sensibles al látex.
04.5 Interacciones con otros medicamentos y otras formas de interacción
La administración concomitante de fondaparinux y sustancias que pueden aumentar el riesgo de hemorragia aumenta el riesgo de hemorragia (ver sección 4.4).
Los anticoagulantes orales (warfarina), los inhibidores plaquetarios (ácido acetilsalicílico), los AINE (piroxicam) y la digoxina no interactuaron con la farmacocinética de fondaparinux. La dosis de fondaparinux (10 mg) en los estudios de interacción fue superior a la dosis recomendada para las indicaciones actuales. Fondaparinux no afecta ni a la actividad INR de la warfarina, ni al tiempo de sangrado en tratamiento con ácido acetilsalicílico o piroxicam, ni a la farmacocinética en estado estacionario de digoxina.
Continuación del tratamiento con otro fármaco anticoagulante.
Si se va a iniciar un tratamiento continuado con heparina o HBPM, como regla general, la primera inyección debe administrarse 1 día después de la última inyección de fondaparinux.
Si es necesario continuar el tratamiento con un antagonista de la vitamina K, debe continuarse el tratamiento con fondaparinux hasta que se alcance el valor de INR establecido.
04.6 Embarazo y lactancia
El embarazo
No hay datos suficientes sobre el uso de fondaparinux en el embarazo. Los estudios en animales son insuficientes con respecto a los efectos sobre el embarazo, el desarrollo embrionario / fetal, el parto y el desarrollo posnatal debido a una exposición limitada. El fondaparinux no debe prescribirse a mujeres embarazadas a menos que sea estrictamente necesario.
Amamantamiento
El fondaparinux se excreta en la leche de rata, pero se desconoce si el fondaparinux se excreta en la leche materna. No se recomienda la lactancia materna durante el tratamiento con fondaparinux, aunque es poco probable que el lactante lo absorba por vía oral.
Fertilidad
No hay datos disponibles sobre el efecto de fondaparinux sobre la fertilidad humana Los estudios en animales no han mostrado ningún efecto sobre la fertilidad.
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas.
04.8 Efectos indeseables
Las reacciones adversas graves notificadas con más frecuencia con fondaparinux son complicaciones hemorrágicas (en varios sitios, incluidos casos raros de hemorragia intracraneal / intracerebral y retroperitoneal) y anemia. Fondaparinux debe usarse con precaución en pacientes que tienen un mayor riesgo de hemorragia (ver sección 4.4).
Se evaluó la seguridad de fondaparinux 2,5 mg en 3.595 pacientes sometidos a cirugía ortopédica mayor de miembros inferiores tratados hasta por 9 días, en 327 pacientes sometidos a cirugía de fractura de cadera tratados durante 3 semanas después de una profilaxis inicial de 1 semana, en 1.407 pacientes sometidos a cirugía abdominal cirugía tratada hasta por 9 días, y en 425 pacientes médicamente relevantes (no sometidos a tratamientos quirúrgicos) con riesgo de complicaciones tromboembólicas tratadas hasta por 14 días.
Las reacciones adversas notificadas por los investigadores como al menos posiblemente relacionadas con fondaparinux se presentan dentro de cada grupo de frecuencia (muy frecuentes ≥1 / 10; frecuentes: ≥ 1/100,
En otros estudios o en la experiencia postcomercialización, se han notificado casos raros de hemorragia intracraneal / intracerebral y retroperitoneal.
Notificación de sospechas de reacciones adversas
Es importante notificar las sospechas de reacciones adversas que se produzcan después de la autorización del medicamento. Permite un seguimiento continuo de la relación beneficio / riesgo del medicamento. Se invita a los profesionales sanitarios a notificar cualquier sospecha de reacciones adversas a través del sistema nacional de notificación. "Anexo V.
04.9 Sobredosis
Las dosis de fondaparinux superiores al régimen recomendado pueden aumentar el riesgo de hemorragia. No se conocen antídotos para el fondaparinux.
La sobredosis asociada con complicaciones hemorrágicas debe implicar la interrupción del tratamiento y la búsqueda de la causa primaria. Se debe considerar el tratamiento apropiado como hemostasia quirúrgica, transfusión de sangre, transfusión de plasma fresco, plasmaféresis.
05.0 PROPIEDADES FARMACOLÓGICAS
05.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: agentes antitrombóticos.
Código ATC: B01AX05.
Efectos farmacodinámicos
Fondaparinux es un inhibidor sintético y selectivo del Factor X activado (Xa). La actividad antitrombótica de fondaparinux es el resultado de la inhibición selectiva del factor Xa mediada por la antitrombina III (ATIII). Mediante la unión selectiva a ATIII, fondaparinux potencia (aproximadamente 300 veces) la neutralización natural del factor Xa por ATIII. El factor Xa interrumpe la sangre cascada de coagulación e inhibe tanto la formación de trombina como el desarrollo de trombos Fondaparinux no inactiva la trombina (Factor II activado) y no tiene efecto sobre las plaquetas.
A la dosis de 2,5 mg, fondaparinux no afecta las pruebas de coagulación de rutina, como el tiempo de tromboplastina parcial activada (aPTT), el tiempo de coagulación activada (ACT) o el tiempo de protrombina (TP) / índice internacional normalizado (INR) en plasma, ni el tiempo de hemorragia o fibrinolítico. Sin embargo, solo se han recibido informes raros de prolongación del aPTT.
Fondaparinux no presenta reacciones cruzadas con suero de pacientes con trombocitopenia inducida por heparina.
Estudios clínicos
Prevención de la tromboembolia venosa (TEV) en pacientes sometidos a cirugía ortopédica mayor de miembros inferiores tratados durante un máximo de 9 días: el plan clínico de fondaparinux se diseñó para demostrar la eficacia de fondaparinux en la prevención de episodios tromboembólicos venosos (TEV), es decir, trombosis proximal y vena profunda distal (TVP) y embolia pulmonar (EP) en pacientes sometidos a cirugía ortopédica mayor de miembros inferiores, como fractura de cadera, cirugía mayor de rodilla o cirugía de reemplazo de cadera. En los ensayos clínicos controlados de fase II y III se estudiaron más de 8.000 pacientes ( fractura de cadera - 1.711, reemplazo de cadera - 5.829, cirugía mayor de rodilla - 1.367). Fondaparinux 2,5 mg una vez al día iniciado 6-8 horas después de la cirugía se comparó con enoxaparina 40 mg una vez al día comenzada 12 horas antes de la cirugía, o 30 mg dos veces al día orno comenzó 12-24 horas después de la cirugía.
En un análisis agrupado de estos estudios, la pauta posológica recomendada de fondaparinux versus enoxaparina se asoció con una disminución significativa (IC 54% -95%, 44%; 63%) en la incidencia de TEV estimada hasta el día 11 después de la cirugía, independientemente del tipo de cirugía realizada. La mayoría de los eventos de "punto final" se diagnosticaron con una venografía predeterminada y se compusieron principalmente de TVP distal, pero la incidencia de TVP proximal también se redujo significativamente. La incidencia de TEV sintomática, incluida la EP, no fue significativamente diferente. Entre tratamientos grupos.
En los estudios versus enoxaparina 40 mg una vez al día iniciados 12 horas antes de la cirugía, se observó hemorragia grave en el 2,8% de los pacientes tratados con fondaparinux a la dosis recomendada en comparación con el 2,6% con enoxaparina.
Prevención de la tromboembolia venosa (TEV) en pacientes sometidos a cirugía por fractura de cadera tratados durante hasta 24 días después de la profilaxis inicial de 1 semana: en un ensayo clínico aleatorizado doble ciego, 737 pacientes fueron tratados con fondaparinux 2,5 mg una vez al día durante 7 ± 1 día después de la cirugía de fractura de cadera. Al final de este período, 656 pacientes fueron aleatorizados para recibir fondaparinux 2,5 mg una vez al día o placebo durante 21 ± 2 días adicionales. Fondaparinux produjo una reducción significativa en la incidencia global de TEV en comparación con placebo [3 pacientes (1,4%) frente a 77 pacientes (35%), respectivamente]. La mayoría (70/80) de los episodios de TEV notificados fueron casos de TVP asintomática detectada flebográficamente El fondaparinux también produjo una reducción significativa en la incidencia de TEV sintomático (TVP y / o EP) [1 (0,3%) frente a 9 (2,7%) pacientes, respectivamente], incluidos 2 EP mortales notificados en el grupo de placebo. Se observó hemorragia grave, todas quirúrgicas y ninguna mortal, en 8 pacientes (2,4%) tratados con fondaparinux 2,5 mg en comparación con 2 (0,6%) con placebo.
Prevención de episodios tromboembólicos venosos (TEV) en pacientes sometidos a cirugía abdominal considerados de alto riesgo de complicaciones tromboembólicas, como pacientes sometidos a cirugía abdominal por cáncer: En un estudio clínico doble ciego, 2.927 pacientes fueron aleatorizados para recibir fondaparinux 2, 5. mg una vez al día o dalteparina 5000 UI una vez al día, mediante una inyección preoperatoria de 2500 UI y una primera inyección postoperatoria de 2500 UI, durante 7 + 2 días. Los sitios principales de cirugía fueron colorrectal, gástrico, hepático, colecistectomía u otras intervenciones biliares. El 69% de los pacientes se sometieron a cirugía por cáncer. Los pacientes se sometieron a cirugía urológica (excluyendo el riñón) o ginecológica. No se incluyeron en el estudio cirugía laparoscópica o vascular. .
En este estudio, la incidencia de TEV total fue del 4,6% (47 / 1.027) con fondaparinux, en comparación con el 6,1% (62 / 1.021) con dalteparina: reducción de la razón impar (IC del 95%) = - 25,8% (-49,7%, 9,5%). La diferencia en la frecuencia de TEV total entre los grupos de tratamiento, que no fue estadísticamente significativa, se debió principalmente a la reducción de la TVP distal. La incidencia de TVP sintomática fue similar entre los dos grupos de tratamiento: 6 pacientes (0,4% ) en el grupo de fondaparinux versus 5 pacientes (0,3%) en el grupo de dalteparina. En el gran subgrupo de pacientes sometidos a cirugía oncológica (69% de la población de pacientes), la frecuencia de TEV fue del 4,7% en el grupo de fondaparinux en comparación con el 7,7% en el grupo de dalteparina.
Se observó hemorragia grave en el 3,4% de los pacientes tratados con fondaparinux y en el 2,4% del grupo tratado con dalteparina.
Prevención de episodios tromboembólicos venosos (TEV) en pacientes médicamente relevantes con alto riesgo de complicaciones tromboembólicas debido a la movilidad reducida durante la enfermedad aguda: En un ensayo clínico aleatorizado doble ciego, se trató a 839 pacientes durante 6 a 14 días con 2,5 mg de fondaparinux una vez al día. o con placebo. Este estudio incluyó pacientes agudos médicamente relevantes de ≥ 60 años que se esperaba que estuvieran postrados en cama durante al menos cuatro días y hospitalizados por insuficiencia cardíaca congestiva clase III / IV de la NYHA y / o enfermedad respiratoria aguda y / o patología aguda infecciosa o inflamatoria. Fondaparinux comparado con placebo redujo significativamente la incidencia global de TEV [18 pacientes (5,6%) frente a 34 pacientes (10,5%), respectivamente]. La mayoría de los eventos fueron TVP distal asintomática. Fondaparinux también redujo significativamente la incidencia de EP considerada fatal [0 pacientes (0,0%) frente a 5 pacientes (1,2%), respectivamente]. Se observó sangrado severo en 1 paciente (0,2%) de cada grupo.
Tratamiento de pacientes con trombosis venosa superficial aguda sintomática espontánea sin trombosis venosa profunda (TVP) concomitante
Un ensayo clínico aleatorizado, doble ciego (CALISTO) incluyó a 3002 pacientes con trombosis venosa superficial espontánea, aguda, sintomática y aislada en miembros inferiores, de al menos 5 cm de longitud, confirmada por ecografía de compresión (CUS). Los pacientes no se incluyeron si tenían TVP concomitante o trombosis venosa superficial dentro de los 3 cm de la unión safeno-femoral. Los pacientes fueron excluidos si tenían insuficiencia hepática grave, insuficiencia renal grave (aclaramiento de creatinina
Los pacientes fueron aleatorizados para recibir fondaparinux 2,5 mg una vez al día o placebo durante 45 días además de medias, analgésicos y / o fármacos antiinflamatorios no esteroideos (AINE) tópicos. El seguimiento continuó hasta el día 77. La población del estudio fue 64% mujeres, con una mediana de edad de 58 años, 4,4% tenía aclaramiento de creatinina.
El resultado primario de eficacia, un resultado compuesto de EP sintomática, TVP sintomática, extensión de la trombosis venosa superficial sintomática, recurrencia de la trombosis venosa superficial sintomática o Muerte en el día 47, se redujo significativamente en un 5,9% en los pacientes del grupo placebo. 0,9% en los que recibieron fondaparinux 2,5 mg (reducción del riesgo relativo: 85,2%; IC del 95%, 73,7% a 91,7% [p
La incidencia de cada componente tromboembólico del resultado primario también se redujo significativamente en los pacientes con fondaparinux como se describe a continuación: EP sintomática [0 (0%) vs 5 (0.3%) (p = 0.031)], TVP sintomática [3 (0.2%) frente a 18 (1,2%); reducción del riesgo relativo 83,4% (p
Las tasas de mortalidad fueron bajas y similares entre los grupos de tratamiento con 2 (0,1%) muertes en el grupo de fondaparinux versus 1 (0,1%) muerte en el grupo placebo.
La eficacia se mantuvo hasta el día 77 y fue constante en todos los subgrupos predefinidos, incluidos los pacientes con venas varicosas y los pacientes con trombosis venosa superficial ubicada debajo de la rodilla.
Se produjo una hemorragia grave durante el tratamiento en 1 (0,1%) paciente con fondaparinux y en 1 (0,1%) paciente con placebo. Se produjo hemorragia no mayor clínicamente relevante en 5 (0,3%) pacientes con fondaparinux y en 8 (0,5%) pacientes con placebo.
05.2 Propiedades farmacocinéticas
Absorción
Después de la administración subcutánea, fondaparinux se absorbe completa y rápidamente (100% de biodisponibilidad absoluta). Tras una única inyección subcutánea de 2,5 mg de fondaparinux a sujetos jóvenes sanos, la concentración plasmática máxima (Cmax media = 0,34 mg / l) se alcanza 2 horas después de la administración. Las concentraciones plasmáticas iguales a la mitad de los valores medios de Cmax se alcanzan 25 minutos después de la administración.
La farmacocinética de fondaparinux es lineal en un rango de dosis de 2 a 8 mg por vía subcutánea en sujetos ancianos sanos. Después de la administración una vez al día, los niveles plasmáticos en estado estacionario se alcanzan de 3 a 4 días después, con un aumento de 1,3 veces en la Cmáx y el AUC.
La media (CV%) de los parámetros estimados en estado estacionario de fondaparinux en pacientes sometidos a cirugía de reemplazo de cadera que recibieron fondaparinux 2,5 mg una vez al día son: Cmax (mg / l) - 0,39 (31%), Tmax (h) - 2,8 (18%) ) y Cmin (mg / l) - 0,14 (56%). En pacientes con fractura de cadera asociada a la vejez, las concentraciones plasmáticas de fondaparinux en estado estacionario son: Cmax (mg / l) - 0,50 (32%), Cmin (mg / l) l) - 0,19 (58%).
Distribución
El volumen de distribución de fondaparinux es limitado (7 - 11 litros). In vitro, fondaparinux se une alta y específicamente a la proteína antitrombina con una unión a la concentración plasmática dependiente de la dosis (98,6% a 97,0% en un rango de concentración de 0,5 a 2 mg / l). Fondaparinux no se une de manera significativa a otras proteínas plasmáticas, incluido el factor plaquetario 4 (PF4).
Dado que el fondaparinux no se une de manera significativa a proteínas plasmáticas distintas de ATIII, no se espera interacción con otros fármacos al cambiar la unión a proteínas.
Biotransformación
Aunque no se ha evaluado completamente, no hay evidencia de metabolismo de fondaparinux y, en particular, de formación de metabolitos activos.
Fondaparinux no inhibe in vitro el sistema CYP450 (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A4). Por lo tanto, no se espera que fondaparinux interactúe en vivo con otros fármacos inhibiendo el metabolismo mediado por CYP.
Eliminación
La vida media de eliminación (t½) es de aproximadamente 17 horas en sujetos jóvenes sanos y de aproximadamente 21 horas en sujetos sanos de edad avanzada El fondaparinux se excreta del 64 al 77% por los riñones como compuesto inalterado.
Categorías especiales de pacientes:
Población pediátrica - Fondaparinux no se ha estudiado en esta clase de pacientes para la prevención de TEV o para el tratamiento de la trombosis venosa superficial.
Pacientes de edad avanzada - La función renal puede disminuir con la edad y, por lo tanto, la capacidad de eliminación de fondaparinux puede verse reducida en los ancianos. En pacientes> 75 años sometidos a cirugía, el aclaramiento plasmático estimado fue de 1,2 a 1,4 veces menor que en pacientes con edad
Insuficiencia renal - En comparación con pacientes con función renal normal (aclaramiento de creatinina> 80 ml / min), el aclaramiento plasmático es de 1,2 a 1,4 veces menor en pacientes con insuficiencia renal leve (aclaramiento de creatinina de 50 a 80 ml / min) y de media 2 veces menor en pacientes. con insuficiencia renal moderada (aclaramiento de creatinina de 30 a 50 ml / min). En insuficiencia renal grave (aclaramiento de creatinina
Género - No hubo diferencia entre los sexos después del ajuste por peso corporal.
Raza - Las diferencias farmacocinéticas debidas a la raza no se han estudiado de forma prospectiva. Sin embargo, los estudios realizados en sujetos sanos asiáticos (japoneses) no revelaron un perfil farmacocinético diferente en comparación con sujetos sanos caucásicos. De manera similar, no se observaron diferencias en el aclaramiento plasmático entre pacientes negros y caucásicos sometidos a cirugía ortopédica.
Peso corporal - El aclaramiento plasmático de fondaparinux aumenta con el peso corporal (aumento del 9% por 10 kg).
Insuficiencia hepática - Después de una dosis subcutánea única de fondaparinux en sujetos con insuficiencia hepática moderada (categoría B de Child-Pugh), la Cmáx y el AUC totales (es decir, unidas y no unidas) disminuyeron un 22% y un 39%, respectivamente, en comparación con los sujetos con función hepática normal. Las concentraciones plasmáticas más bajas de fondaparinux se han atribuido a la unión reducida a ATIII, que a su vez depende de las concentraciones plasmáticas más bajas de ATIII en sujetos con insuficiencia hepática, lo que, por lo tanto, da como resultado un aumento en el aclaramiento renal de fondaparinux. Se espera que las concentraciones de fondaparinux libre permanezcan sin cambios en pacientes con insuficiencia hepática leve o moderada y, por lo tanto, no es necesario ajustar la dosis en función de la farmacocinética.
05.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y toxicidad.Los estudios en animales son insuficientes con respecto a los efectos de toxicidad reproductiva debido a la exposición limitada.
06.0 INFORMACIÓN FARMACÉUTICA
06.1 Excipientes
Cloruro de sodio
Agua para preparaciones inyectables
Ácido clorhídrico
Hidróxido de sodio
06.2 Incompatibilidad
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
06.3 Período de validez
3 años.
06.4 Precauciones especiales de conservación
Conservar por debajo de 25 ° C. No congelar.
06.5 Naturaleza del envase primario y contenido del envase.
Vidrio tipo I (1 ml) equipado con una aguja de calibre 27 x 12,7 mm y bloqueado por un sistema de bloqueo de pistón de elastómero de bromobutilo o clorobutilo.
Arixtra está disponible en envases de 2, 7, 10 y 20 jeringas precargadas. Hay dos tipos de jeringas:
• jeringa con pistón amarillo y con sistema de seguridad automático
• jeringa con émbolo amarillo y sistema de seguridad manual.
Es posible que no se comercialicen todos los tamaños de envases.
06.6 Instrucciones de uso y manipulación
La inyección subcutánea se administra como con una jeringa convencional.
Las soluciones parenterales deben examinarse visualmente antes de la administración para detectar partículas anormales y tinción.
Las instrucciones para la autoadministración se encuentran en el prospecto.
El sistema de protección de la aguja de las jeringas precargadas de Arixtra ha sido diseñado con un sistema de seguridad para proteger contra pinchazos accidentales con la aguja después de la inyección.
Los medicamentos no utilizados y los desechos derivados de este medicamento deben eliminarse de acuerdo con las regulaciones locales.
07.0 TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Aspen Pharma Trading Limited
3016 Lake Drive
Campus de negocios de Citywest
Dublín 24
Irlanda
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU / 1/02/206 / 005-008
035606060
035606072
EU / 1/02/206/024
EU / 1/02/206/025
035606223
EU / 1/02/206/026
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 21 de marzo de 2002
Fecha de la última revalidación: 21 de marzo de 2007
10.0 FECHA DE REVISIÓN DEL TEXTO
D.CCE Agosto de 2014






















.jpg)